(346k) Leveraging Quantum-Chemical Screening Methods to Guide the Discovery of Promising Metal?Organic Frameworks
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Computational Molecular Science and Engineering Forum
Poster Session: Computational Molecular Science and Engineering Forum (CoMSEF)
Wednesday, November 18, 2020 - 8:00am to 9:00am
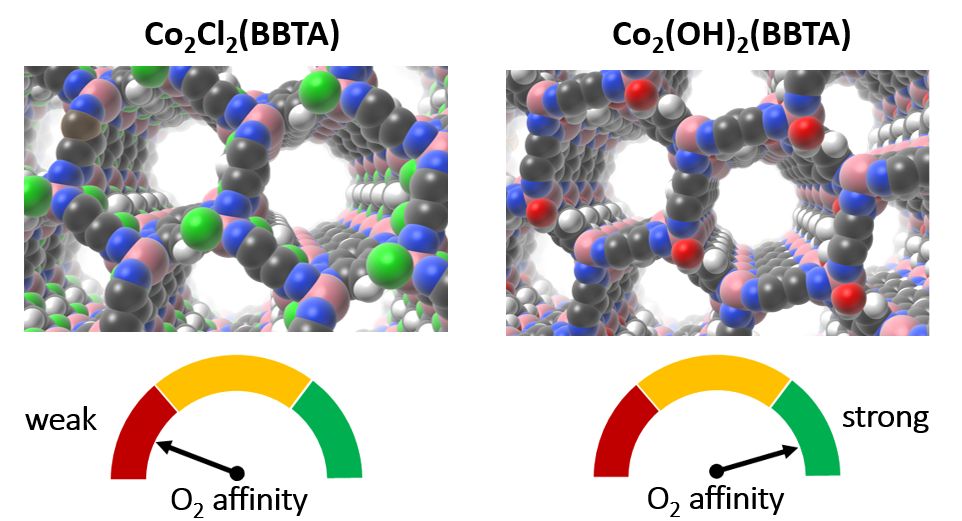
Recently, we developed a high-throughput periodic DFT workflow to computationally investigate large numbers of MOFs in a robust and automated manner, with a specific focus on catalytic and gas separation applications involving the binding and activation of small molecule adsorbates. As one representative example, we highlight how this workflow has been used to identify a simple ligand-exchange procedure that can turn an otherwise unselective cobalt MOF into one that strongly and selectively binds O2 over N2, which is appealing for industrial air separation applications. Experimentally obtained adsorption isotherms confirm this finding and indicate that the identified material has one of the highest room-temperature O2/N2 selectivities of a MOF to date. More broadly, we demonstrate how our automated periodic DFT workflow has been used to construct the largest database of MOF quantum-chemical properties and the implications that this may have for materials design in the future. We briefly discuss our ongoing efforts to leverage this resource to predict the quantum-chemical properties of tens of thousands of MOFs in a matter of seconds via recent advances in machine learning.