(398i) Bridging Mechanism of V2+/V3+ Reaction on Glassy Carbon in Acidic Electrolytes for Vanadium Redox Flow Batteries
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Transport and Energy Processes
Advanced Electrochemical Energy Storage Technologies
Wednesday, November 18, 2020 - 9:45am to 10:00am
Redox Flow Batteries (RFBs) have shown tremendous capability to store and release renewable electricity at a multimegawatt-hours scale with long electrode lifetimes. RFBs have a unique ability to decouple energy and power which provides them high scalability and flexibility for grid-energy storage applications. Among aqueous RFBs, all-vanadium RFBs (VRFBs: VO2+/VO2+//V2+/V3+) are among the most developed RFBs and the closest to commercial implementation. However, VRFBs suffer from a high kinetic overvoltage of the slower V2+/V3+ redox couple (V2+ ⇄ V3+ + e−, V vs SHE) that lowers the overall battery efficiency.1 Despite substantial work on VRFBs, there is a lack of fundamental understanding of V2+/V3+ redox couple chemistry, particularly the role of electrolyte and the associated charge transfer. VRFBs with carbon electrodes employing hydrochloric acid (HCl) or mixed acid electrolytes (HCl/H2SO4)1 have higher current densities compared with the traditionally used sulfuric acid (H2SO4) electrolyte, but it is unclear if the enhancements are due to kinetics, mass transport, or conductivity.
Recently, we have reported V2+/V3+ kinetic measurements on glassy carbon in HCl, H2SO4, and mixed HCl/H2SO4 electrolytes for VRFBs. We extract exchange current densities (io) via steady-state current measurements (io,Tafel) and impedance spectroscopy (io,Rct) and apparent activation energies (Ea). Our results from both independent techniques to measure io show that the V2+/V3+ reaction kinetics in HCl are ~2.5 times faster than in H2SO4 or HCl/H2SO4. By comparing values of io,Rct to io,Tafel, we also show that V2+/V3+ reaction is an overall two electron transfer reaction in HCl (with one electron in rate determining step, RDS), as opposed to an overall one electron transfer reaction in H2SO4 and HCl/H2SO4. We propose that the enhanced V2+/V3+ redox kinetics in HCl are caused by the charge transfer proceeding through more polarizable chloride (*Cl) bridge, instead of through surface-bound hydroxyl (*OH) groups in H2SO4 and HCl/H2SO4.2 We further hypothesize that the anion’s bridging strength, which is a measure of deformability in the presence of electric field,3 will govern the V2+/V3+ reaction kinetics, with stronger bridging strength corresponding to higher activity.
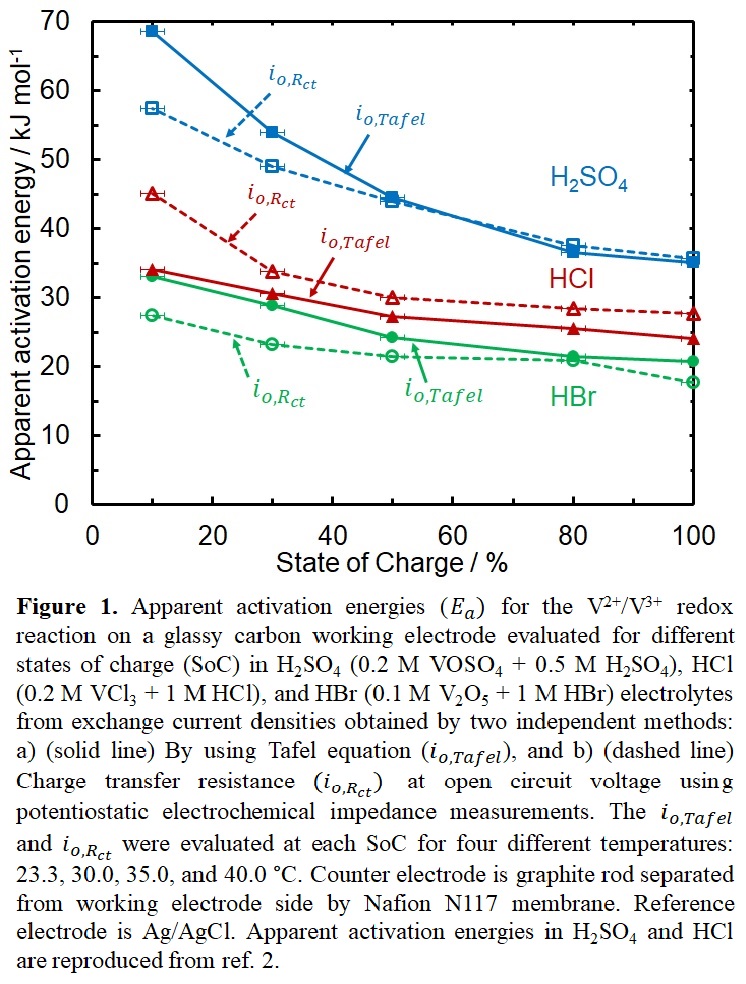
In this talk, we will discuss our V2+/V3+ kinetic measurements on glassy carbon in hydrobromic (HBr) and hydriodic (HI) acid to test the hypothesis of bridging strength as the sole criteria governing V2+/V3+ reaction kinetics. The bridging strength of anions follows the order, I > Br > Cl > OH,3 so based on our hypothesis we expect the V2+/V3+ reaction rates to follow the same trend. The V2+/V3+ exchange current densities in HBr are ~1.5 times higher than in HCl, with 5 to 15 kJ mol-1 lower Ea (Figure 1). The io increases and the Eadecreases with increase in State of Charge (SoC) in HBr, similar to the behavior observed in HCl and H2SO4. We determine that the reaction in HBr is an overall two electron transfer reaction, with one electron involved in RDS, similar to HCl. Further, the V2+/V3+ reaction is HI has kinetics faster than HBr, with a maximum rate at ~50% SoC. The io,Rct in HI indicates the reaction mechanism changes to an overall one electron transfer. However, we notice that the rates in HI are affected immensely by the presence of small amount of sulfate anions. This points to the bridging strength not being a sole descriptor for the reaction kinetics. We attribute this observed trend in rates of V2+/ V3+ reaction to be dependent on the stability of the vanadium intermediate formed on the surface of glassy carbon, computed using density functional theory.
REFERENCES:
- Choi, C. et al. Renew. Sustain. Energy Rev. 69, 263–274 (2017).
- Agarwal, H. et al. ACS Energy Lett. 4, 2368–2377 (2019).
- Anbar, M. et al. J. Phys. Chem. 69, 973–977 (1965).