(3ab) Integrated Materials Engineering: Material Design and Development Combined with Multi-Scale Computational Modeling and Artificial Intelligence for Energy and Healthcare
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Meet the Candidates Poster Sessions
Meet the Faculty and Post-Doc Candidates Poster Session
Monday, November 16, 2020 - 8:00am to 9:00am
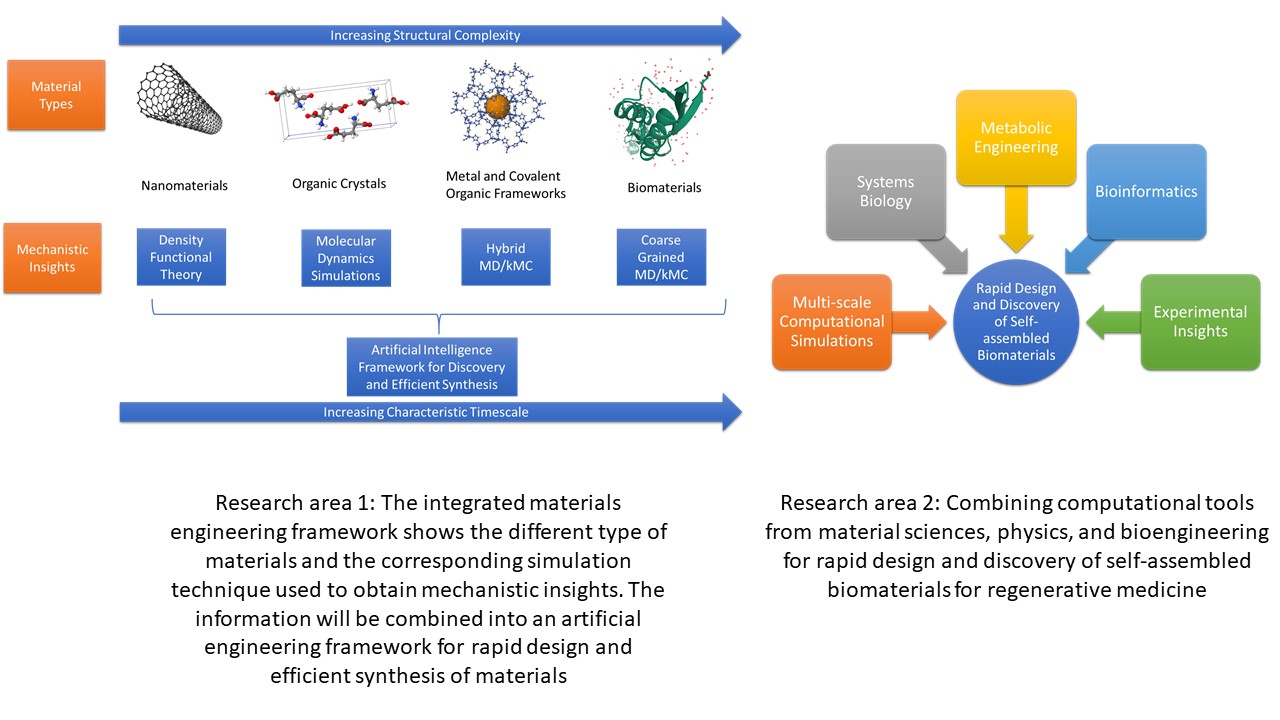
Research Area 1: Multi-scale Computational Modeling Coupled with Artificial Intelligence for Design and Development of Self-assembled Materials
Description: Computational modeling is one of the critical components in the field of material sciences. The aim of using computational techniques is to identify the crucial molecular events which can help in discovery and design of the materials. At the beginning, the computational techniques were limited to explore the different states of the system near the local minima or near equilibrium of different states. More recently, the computational techniques are being evolved to capture the non-equilibrium dynamics of the system which govern the properties of the materials. Different computational techniques are effectively combined to capture the non-equilibrium dynamics which usually spans multiple time and length scales. However, most multi-scale computational models are specific to systems being studied and a more robust framework combined with artificial intelligence needs to be developed. Such framework can unravel the relationship between the key molecular events and the material properties leading to smarter and more efficient synthesis techniques. For example, the crystallization of organic molecule is governed by solvation shell dynamics and the crystallization of Metal Organic Frameworks (MOFs) is governed by the electron exchange between metal ligand and organic linker. However, the solvation shell dynamics of organic linker during MOF synthesis can also affect the electron exchange in MOF crystallization. In order to facilitate higher electron exchange during the MOF synthesis process, the insights gained from computational models of organic crystallization can be used to design solvent during MOF synthesis to reduce the solvation shell effects and allow greater interaction between the components. It shows that integrated development of materials can reduce time for design and discovery of novel materials. This phenomenon has also been captured in Material Genome Initiative (MGI) by National Science Foundation (NSF). The MGI strategic plan has stressed the importance of creating accurate and reliable simulations and the data analytics to enhance the value of the computational and experimental data. With the experience I have gained in using multitude of different computational techniques during my PhD, I plan to create an integrated environment of the materials engineering, which will gather the data from previous experimental and computational studies, fill in the gaps of knowledge using appropriate computational or experimental technique and accelerate the design and development of novel materials using machine learning and artificial intelligence.
The proposed research plan could significantly improve the design and synthesis of the metal-organic frameworks (MOFs) and covalent organic frameworks (COFs). These organic frameworks have highly symmetric, porous crystalline structure. This property allows them to be used for energy storage and transport. Currently, there are few thousand of MOFs and COFs have been theoretically designed. However, less than fifty of those have been experimentally synthesized. The reproducibility of the published results lies somewhere between 6-10%. Inefficient mechanistic analysis, inconsistencies in reporting results, lack of comprehensive databases of experimental and computational studies and variability of energy calculation methods are some of the main challenges faced by the researchers.
To overcome the limitations, it necessary to gain mechanistic insights into the self-assembly by capturing non-equilibrium dynamics. Current state-of-the-art multi-scale non-equilibrium simulation techniques for such complex materials include computationally intensive energy exploration methods using various sampling methods. Uniform sampling of the system variables, energy calculation and knowledge of end states of the system are the important challenges in these types of calculations. Performing such techniques on every system for design and discovery of materials could require decades of computational power. The proposed research plan would help reduce these bottlenecks as follows:
- Perform non-equilibrium mechanistic modeling to gain mechanistic insights into self-assembly
- Create a database of experimental and computational results to gain mechanistic insights and establish a strong communication between experimental and computational research field
- Fill in the knowledge gaps as much as possible using the necessary computational or experimental methods
- Apply the machine learning methods to identify the impactful variables and similarities in the mechanism of self-assembly of different types of materials
- Create an artificial intelligence framework to accelerate the design and development of the materials
Although the research area is inspired from the progress of MOFs and COFs, the methodology is applicable to many different types of self-assembled materials including nanomaterials, biomaterials, polymers etc. The robustness of the proposed plan can help solve some of the most challenging problems of the current century and will promote strong collaboration with different research fields.
Research area 2: Integrated Computational Modeling of Self-assembled Biomaterials for Regenerative Medicine
Description: The demand for tissue and organ replacement is ever increasing. However, the complexity of biological environment, expensive and long period of clinical trials prevents rapid development of new biomaterials for regenerative medicine. The field of biomaterials has great overlap with chemical engineering, material science, biology, physics, and mathematics. For effective and rapid design and discovery of biomaterials it is necessary to bring the synergies of engineering tools such as multi-scale molecular modeling, systems biology, metabolic engineering and the “omic sciences†such as (genomics, proteomics, metabolomics etc.) in one robust framework. Furthermore, the biomaterials which can self-assemble are most desired since they allow non-invasive solutions for regenerative medicine. For this reason, the robust framework discussed earlier is applicable in the field of self-assembled biomaterials for regenerative medicines.
To approach such highly interdisciplinary field, at first it is important to formulate a framework of simulation technique which can combine the multiscale molecular simulations from material sciences and combine with the computational tools developed for biological systems analysis. Current state-of-the-art techniques of analysis are focused on only few of the aspects of the biological systems. The proposed research plan includes:
- Formulation of simulation scheme which combines principles from materials sciences and biological systems analysis
- Identify the important variables
- Create a database of the experimental computational results
- Apply the principles of machine learning and artificial intelligence to accelerate the discovery and design of self-assembled biomaterials for regenerative medicine
The interdisciplinary approach and emphasis on mechanistic understanding of the system are the most important features described in the proposed research plan. The proposed research plan is entirely in-tune with the MGI of NSF.
Teaching Interests:
Creating a strong fundamental understanding of the research and motivating students to pursue graduate studies are two of the most important aspects of teaching. It develops the next generation of students to perform cutting edge research and create different industries. For this purpose I have designed the courses on Mathematical Methods in Chemical Engineering, Transport Phenomena and Algorithms in Chemical Engineering. Mathematical methods and transport phenomena are based on the traditional Chemical Engineering principles to strengthen the fundamentals. Each of the course module includes a case study which relates the classroom understanding with the real-world problems. The course in Algorithms in Chemical Engineering will allow students to develop their own code for the computational modeling. It will cover different computational techniques such as molecular dynamics (MD), kinetic Monte-Carlo (kMC), and systems analysis.
During my PhD, I have always scored above 4.25 out of 5 in the post-semester instructor survey. Furthermore, UIC has a diverse student body which has improved my communication skills to understand the concerns of the students. By holding highest standards of teaching, I plan to create a competitive next generation of students.