2020 Virtual AIChE Annual Meeting
(433i) Deposition and Structure of MoO3 Clusters on Anatase TiO2(101)
Authors
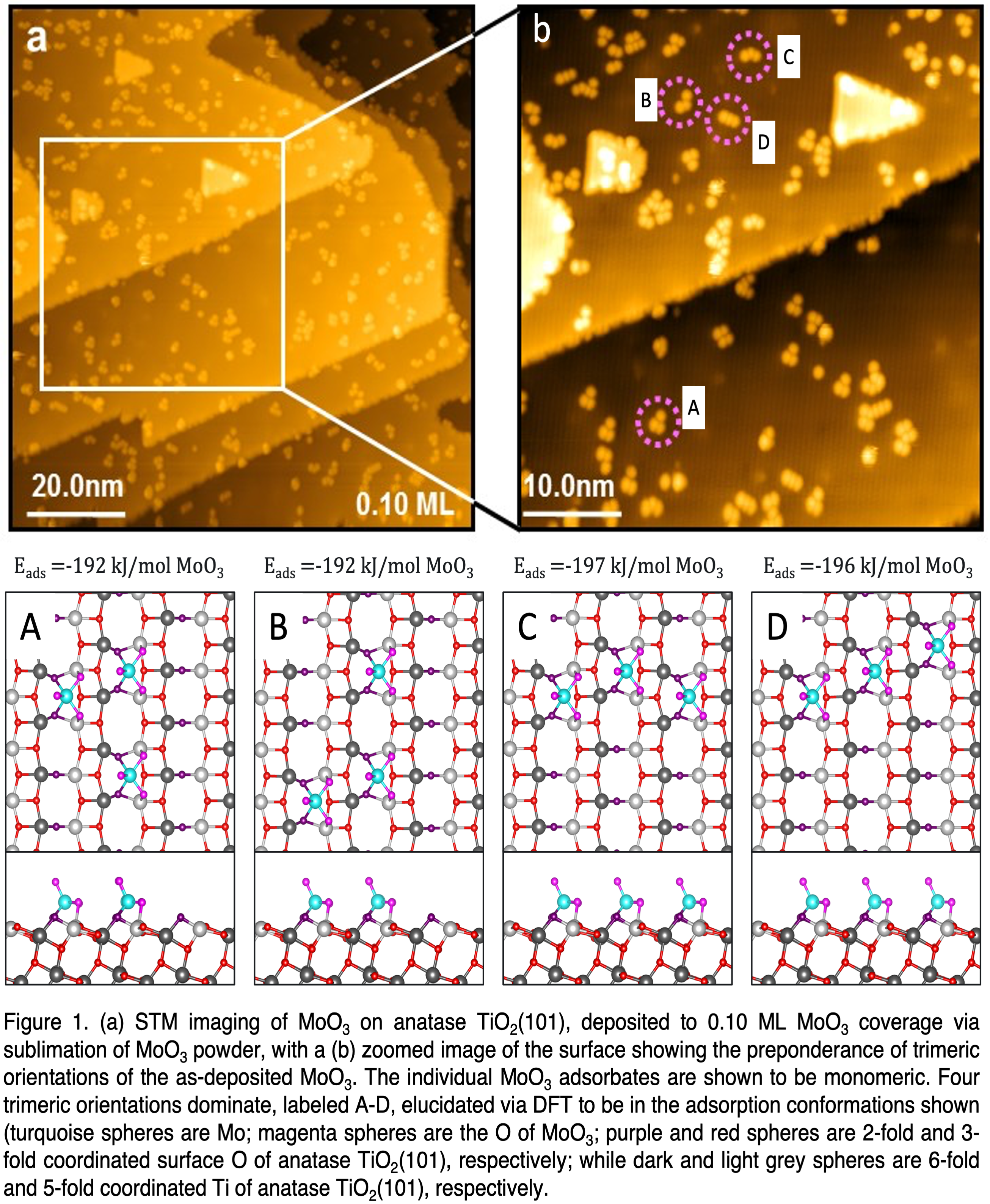
Monodispersed MoO3 clusters are deposited by the sublimation of MoO3 powder at 1350 K over the anatase TiO2(101) surface. Since the stable gas phase structure of MoO3 is a trimer in cyclic configuration, i.e. (MoO3)3,[1, 4] it is assumed that MoO3 deposits as this structure. After deposition, the STM images of the lowest concentration of MoO3 show no preference for step edges, but three-unit clustering is observed. The adsorbed clusters appear as bright protrusions (see Figure 1a), with an apparent cluster height of approximately 1.5 Å. Annealing to 450 K results in a better-ordering of the overlayer, but further annealing to 650 K leads to three-dimensional clusters. The XPS results indicate that the Mo(3d5/2) binding energy in as-deposited (MoO3)3 is characteristic of Mo6+, and the oxidation state of Mo remains (+6) upon heating to 500 K, consistent with DFT results suggesting that the O of MoO3 monomers are very tightly bound.
AIMD simulation of the most stable adsorption configuration of terrace-bound (MoO3)3 reveals that this structure quickly decomposes into monomeric units of MoO3. This decomposition is shown to be primarily entropically driven. As can be seen in Figure 1(a-b), STM imaging clearly shows that these monomeric units preferentially remain in one of four as-decomposed trimeric orientationsâthe three-unit clusters seen in STMâelucidated via DFT to be isoenergetic structures labeled A-D in Figure 1. DFT calculations show isolated monomers to be energetically (and naturally, entropically) preferred over these trimeric orientations. However, activation barriers for monomer MoO3 diffusion are calculated to be significant. The along-row diffusion barrier is shown to be thermally surmountable while the across-row diffusion barrier is prohibitively high. This effectively constrains MoO3 monomers to remain in the A-D trimeric orientations provided high temperatures are not accessed, at which point they are constrained to a single anatase TiO2(101) row, similar to that found for adsorbed H.[5] As such, this system may offer great promise as an ideal platform for reactivity studies on well-defined supported model transition-metal oxide catalysts.
References:
- Rousseau, R., et al., Chemical Society Reviews, 2014. 43(22): p. 7664-7680.
- Bondarchuk, O., et al., Angewandte Chemie International Edition, 2006. 45(29): p. 4786-4789.
- Tang, X., et al., Journal of Physics: Conference Series, 2013. 438: p. 012005.
- Berkowitz, J., M.G. Inghram, and W.A. Chupka, The Journal of Chemical Physics, 1957. 26(4): p. 842-846.
- Doudin, N., et al., ACS Catalysis, 2019. 9(9): p. 7876-7887.