(606d) Molecular Simulation of CO2 Sorption Effects on Structure and Local Dynamics of Polystyrene Melts
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Materials Engineering and Sciences Division
Polymer Simulations: Molecular Structure and Function
Friday, November 20, 2020 - 8:30am to 8:45am
Generation of realistic initial polymer structures at high Mw is not feasible by conventional methods, such as Molecular Dynamics (MD). In these cases, a hierarchical scheme was used for the generation of the polymer systems, which consisted of equilibration at a coarse-grained level of representation through efficient connectivity-altering Monte Carlo simulations,[1] and reverse-mapping back to the atomistic representation,[2] obtaining the configurations used for subsequent MD simulations. Low Mw polymeric chains were generated and equilibrated directly at the atomistic level.
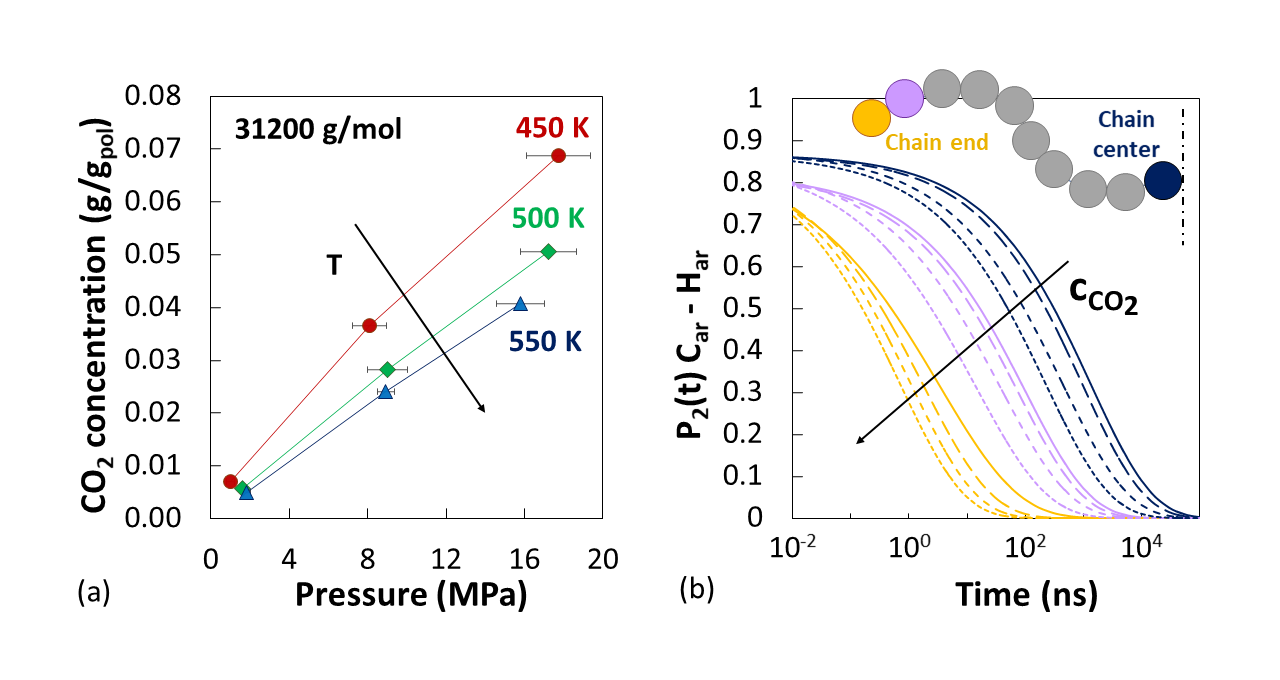
Sorption isotherms and associated swelling effects were determined using an iterative scheme[3] that incorporated MD simulations in the NPT ensemble and the Widom particle insertion method, to ensure consistency between CO2 fugacity in the gas phase and in the gas-polymer mixture. CO2 diffusion coefficients were extracted from the Mean Square Displacement of the gas molecules during long MD runs in the NVE ensemble. Solubility and diffusivity compared well with experimental results and with predictions of the Sanchez-Lacombe equation of state, which were obtained for comparison where experimental data were not available. Chain dimensions and structural features of the polymer matrix, such as radial distribution functions and x-ray scattering patterns, were analysed and found to be in close agreement with experimental data. In the case of gas polymer systems, it was found that CO2 affects interchain packing more significantly than the average chain dimensions. The segmental dynamics of the polymer was evaluated, in terms of time autocorrelation functions of several characteristic vectors of the polymer. In the presence of CO2, a significant acceleration of the segmental dynamics of the polymer occurred, more pronouncedly at low Mw. The speed-up effect caused by the swelling agent was not limited to the chain ends but affected the whole chain in a similar fashion.
References:
[1] Vogiatzis G. G., Theodorou D. N., 2104. Local segmental dynamics and stresses in polystyrene-C60 mixtures, Macromolecules, 47, 387-404.
[2] Ndoro T., Voyiatzis E., Ghanbari A., Theodorou D. N., Böhm M. C., Müller-Plathe F., 2011. Interface of Grafted and Ungrafted Silica Nanoparticles with a Polystyrene Matrix : Atomistic Molecular Dynamics Simulations, Macromolecules, 44, 2316-2327.
[3] Spyriouni T., Boulougouris G.C., Theodorou D.N., 2009. Prediction of sorption of CO2 in glassy atactic polystyrene at elevated pressures through a new computational scheme Macromolecules, 42, 1759 – 1769.
Figure. (a) Simulated sorption isotherms of CO2 in aPS at different temperatures. (b) Acceleration of the dynamics of the C–H bonds on the aromatic rings of aPS (Car–Har) due to increasing CO2 concentration (shorter dash lines) in the simulated systems. The colors indicate different positions along the chain.