(631g) Mapping Catalytic Reaction Pathways through Combined Theory and Surface Sensitive Spectroscopy: Decomposition of Phenolics on Pt(111) As a Case Study
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Catalysis and Reaction Engineering Division
Fundamentals of Catalysis and Surface Science II: Descriptors and Trends in Reactivity
Wednesday, November 18, 2020 - 9:30am to 9:45am
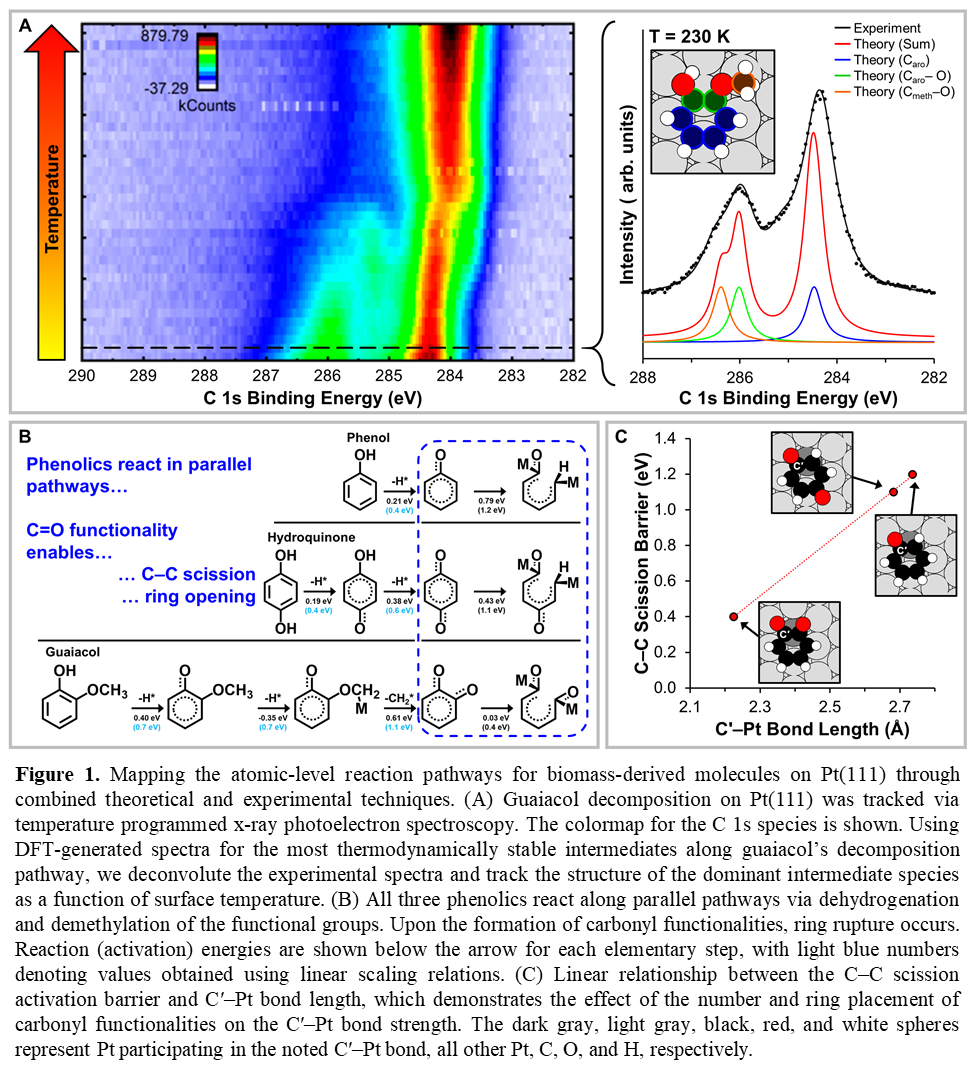
Here, we demonstrate the efficacy of one such approach by mapping the atomic-level reaction pathways for the thermal decomposition of biomass-derived phenolic compounds (i.e. phenol, hydroquinone, and guaiacol) on Pt(111). Each species’ C 1s and O 1s binding energies are measured via synchrotron-based x-ray photoelectron spectroscopy as a function of surface temperature (Figure 1A, left). By combining deconvolution analyses of the experimental spectra with DFT-generated spectra for the most energetically favorable phenolic intermediate species (Figure 1A, right), we identify temperature dependent structural changes in the adsorbed species and, thus, track the atomic-level conversion of complex molecules during the reaction.
Our approach reveals that the decomposition for three model phenolic compounds follow parallel pathways (Figure 1B). The reaction begins by sequential dehydrogenation and demethylation of the functional groups. Resulting carbonyl functionalities disrupt the ring structure’s aromaticity, weakening the C–C bonds and enabling ring rupture. The number and relative orientation of the carbonyl functionalities alters the C–C scission activation barrier (Figure 1C) by withdrawing charge density from the aromatic ring. Compensating charge density transfer from the Pt surface consequently strengthens C–Pt bonds (Figure 1C), resulting in C–C scission. Overall, our work provides a blueprint for the integration of theoretical calculations and surface sensitive spectroscopy in mapping complex reaction pathways.