(105b) Machine Learning-Enabled Optimization of Classical Molecular Models
AIChE Annual Meeting
2022
2022 Annual Meeting
Computing and Systems Technology Division
CAST Director's Student Presentation Award Finalists (Invited Talks)
Monday, November 14, 2022 - 12:45pm to 1:00pm
As an example, we investigate harnessing machine learning, data science, and optimization techniques commonly used by the process systems engineering community to address a long-standing challenge within the molecular modeling and simulations community: efficiently calibrating molecular models. Molecular simulation, which is a powerful tool for studying the thermodynamic and dynamic properties of materials, can be used within our integrated multiscale modeling and design framework to screen vast molecular design space and optimize resource-intensive experimental characterization. With progress made by recent molecular engineering initiatives, including designer solvents, such as ILs, porous materials, and biological systems, as well as advances in computational power, molecular simulation capabilities have rapidly progressed. However, using molecular simulation in a quantitative and predictive capacity as is necessary for its use as a data generator and high-throughput screening tool within a data-to-design workflow requires accurate molecular models, called force fields. Developing force fields is a laborious, time-consuming endeavor due to the expense of calculating the objective function, or error between the simulation output and the property the simulation is trying to capture. Though off-the-shelf force fields offer accurate predictions for some systems, they lack quantitative accuracy across the extraordinary range of chemistries found in the natural and synthetic world. Thus, further manual parameter tuning, an effort which often takes months, is standard practice to ensure the model has the required accuracy for the molecule(s) and properties of interest [6]. The lack of tools to rapidly calibrate force field models represents a barrier to innovation enabled by predictive modeling.
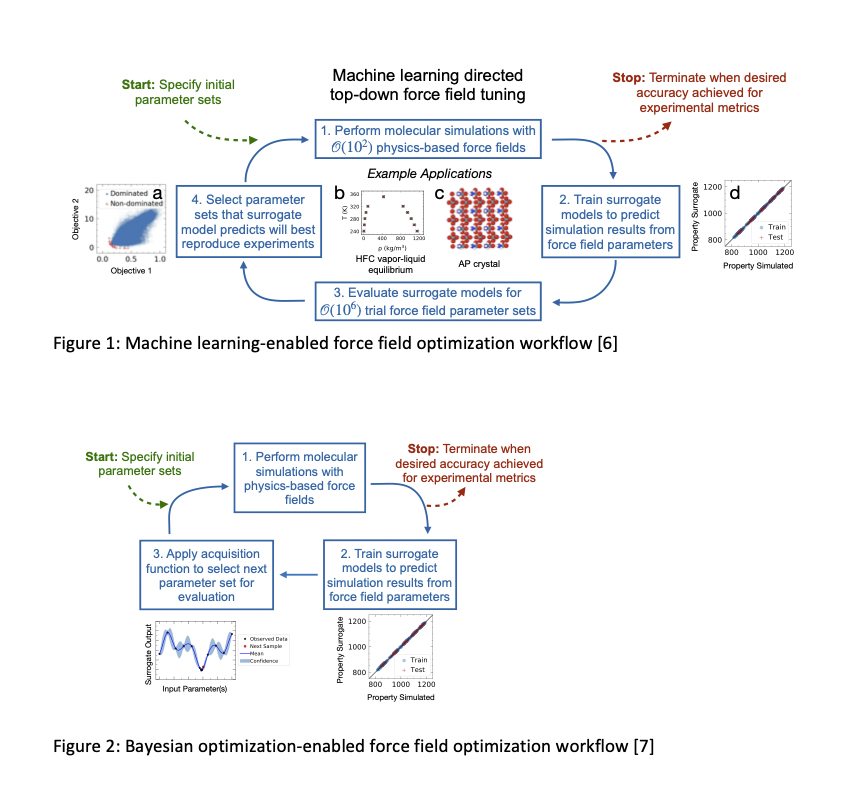
Thus we propose a machine learning-enabled force field optimization framework (see Figure 1). While force field development can be achieved by fitting parameters to data from quantum calculations, here we show surrogate assisted optimization, integrating Gaussian Process regression models with Bayesian optimization, facilitates rapid “top-down†calibration of van der Waals interaction parameters within a force field to reproduce experimental property measurements. This calibration workflow is framed as a multi-objective optimization scheme and we utilize dominated versus non-dominated criteria to find multiple parameter sets with low error in reproducing multiple thermophysical properties. This work challenges existing ideas within the molecular modeling community that an objective “best†force field exists. We show the effectiveness of using surrogate models for force field calibration, establishing that surrogate models can identify accurate force fields using limited training data. This is because the physics-based nature of force fields, including physically interpretable parameters and functional forms, makes them extensible to multiple properties, including those to which they were not calibrated [6]. We compare the efficiency of parameter space exploration and exploitation using iterative surrogate model updates enabled through large samples of parameter space with automated Bayesian sampling (see Figure 2). We find that Bayesian optimization finds lower error parameter sets in half the time [7]. As a demonstration case, we show these methods provide a quick and efficient route to optimized molecular models which reproduce vapor-liquid equilibrium (VLE) properties for two environmentally relevant hydrofluorocarbons, HFC-32 and HFC-125.
Finally, we evaluate the insights into classical molecular modeling paradigms and the guidance these tools provide to experimental and process modeling collaborators within the data-to-design framework. Because we found multiple low error parameter sets for each HFC that were well-spread out through parameter space, we performed analyses using traditional applied statistics techniques to understand the apparent over-parameterization of the system. For example, we performed a local identifiability analysis and showed that the force field models for both HFCs were not fully identifiable when tuning to a single property (liquid density), but became so when full VLE data were included in the calibration workflow, indicating the importance of measuring a variety of data and including uncorrelated properties in an optimization procedure. Additionally we investigate using model selection techniques while simultaneously calibrating multiple HFC force fields to determine the number of parameters which balance the complexity of the model optimization procedure with the quality of fit. We perform preliminary efficiency analyses of the formulation of the Bayesian optimization workflow to determine the most effective method for automatically finding optimal parameter sets. As further capabilities are developed, this tool will be harnessed to rapidly calibrate molecular models for other systems of interest and multiscale design schemes to facilitate rapid molecular and process design and optimization. We emphasize the significance of this work in the molecular modeling community, as rapid generation of accurate molecular models is the key to harnessing the full quantitative predictive capacity of molecular simulations.
References
[1]United Nations Environment Programme. Ozone Secretariat. (2006). Handbook for the Montreal protocol on substances that deplete the ozone layer. UNEP/Earthprint.
[2]EPA, Phasedown of hydrofluorocarbons: Establishing the allowance allocation and trading program under the American innovation and manufacturing act (DoA: 2021).
[3]Plechkova, N. V., & Seddon, K. R. (2008). Applications of ionic liquids in the chemical industry. Chemical Society Reviews, 37(1), 123-150.
[4]Chávez-Islas, L. M., Vasquez-Medrano, R., & Flores-Tlacuahuac, A. (2011). Optimal molecular design of ionic liquids for high-purity bioethanol production. Industrial & Engineering Chemistry Research, 50(9), 5153-5168.
[5]Holbrey, J. D., & Seddon, K. R. (1999). Ionic liquids. Clean products and processes, 1(4), 223-236.
[6]Befort, B. J., DeFever, R. S., Tow, G. M., Dowling, A. W., & Maginn, E. J. (2021). Machine learning directed optimization of classical molecular modeling force fields. Journal of Chemical Information and Modeling, 61(9), 4400-4414.
[7]Bridgette J. Befort, Ryan S. DeFever, Edward J. Maginn, Alexander W. Dowling. Machine Learning-Enabled Optimization of Force Fields for Hydrofluorocarbons (2021). PSE2021+. Accepted for publication.