2022 Annual Meeting
(191h) Bridging Thermal and Electrochemical Catalysis: Rational Catalyst Design at Atomic Scales
Author
In this talk, I will discuss how a wide range of computational methods can accelerate catalyst discovery. In particular, I will show how combining physical models, high-throughput simulations, density functional theory (DFT), and machine learning (ML) can elucidate different aspects of materials discovery and catalyst design in thermal and electrocatalysis.
I firstly illustrate the case for building an understanding of surface restructuring and sintering in heterogeneous catalytic contexts since the durability and morphology/shapes of active metal sites under reaction conditions are areas of great significance for catalyst design. For this purpose, I build a training set of simple first-principle DFT calculations of activation energies for atomic diffusions/migrations over various metal FCC surfaces. From these, I then extract coordination-based parameters to fit to the energies of atoms at non-equilibrium distances. Such extension of our previously developed coordination-based schemes [1] breaks down any sintering process into individual atomic diffusions such that they can be fed to a kinetic Monte Carlo (kMC) approach for modeling the migration of nanoparticles on various supports through random fluctuations in the nanoparticle structure [2]. This can be then compared with experimental sintering patterns. Since surface energies and the distribution of site morphologies are vital for optimization of catalytic activity and selectivity, using mentioned coordination schemes with structural and site information, we also evaluate surface energies and predict relative stabilities of various catalytic surfaces with periodic slabs as well as various metal nanoparticle (MNP) shapes [3]. Finally, we also propose to extend these models to multimetallic surfaces (alloys). This can lead to effective screening of thermodynamic stabilities of alloy MNPs. As such, the resulting physical model can be applied to calculate the energetics of any nanoparticle morphology and chemical composition, thus significantly accelerating design of durable nanoalloys.
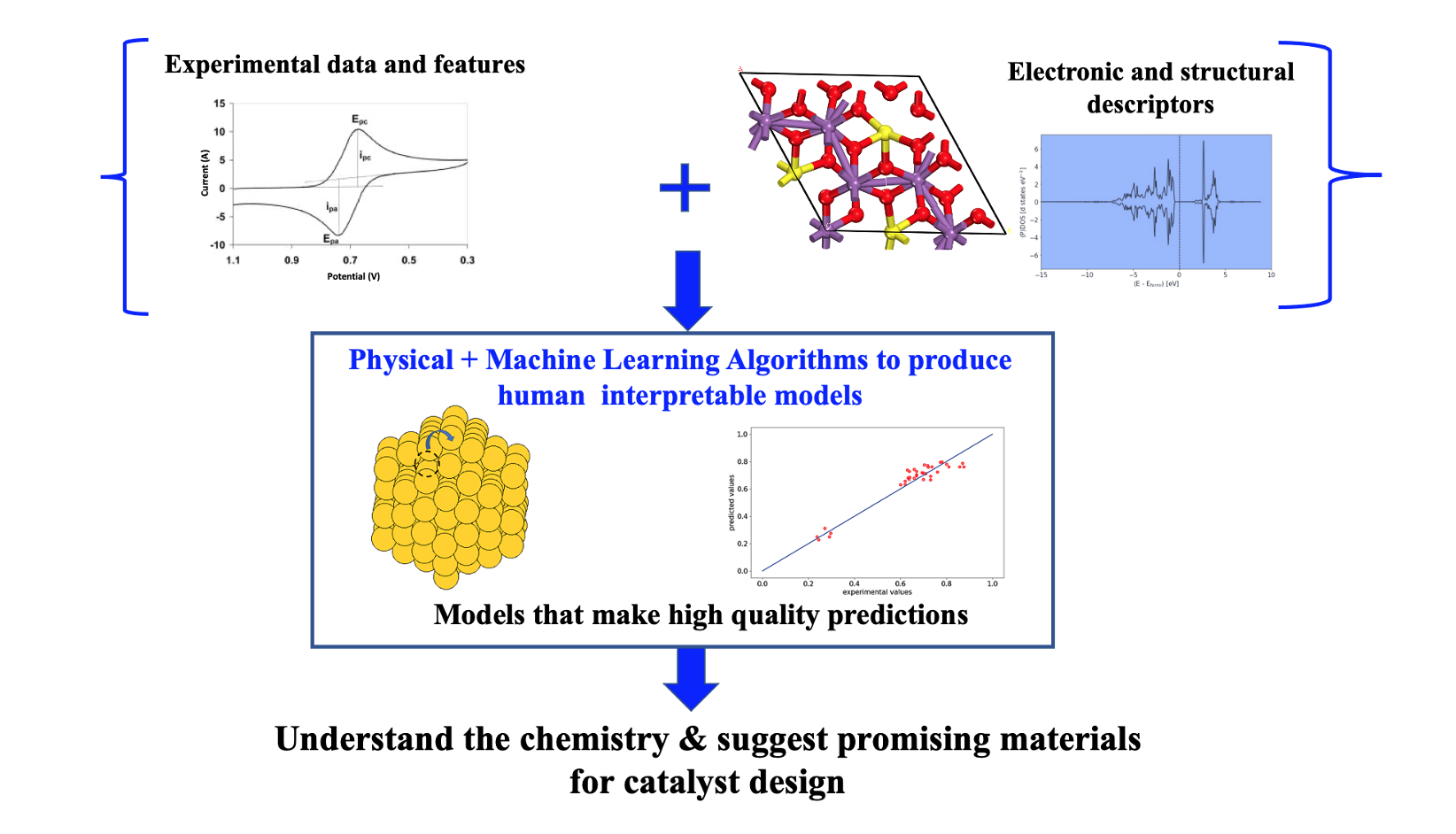
In the second example, we employ a high-throughput exploration and computation of unexplored materials of high interest for OxR, and their relevant properties (for the respective reactions), based on a large number of bulk prototype materials including antimonates, oxides, pyrochlores, alloys, nitrides, and sulfides. Such effort is critical for broader, efficient use of fuel cell and water-splitting devices. The methodology leverages prototype DFT calculations to extract electronic and structural descriptors from bulk crystal structures of materials, and then, employ ML approaches to efficiently arrive at the right set of descriptors for making predictions under catalytically relevant conditions. Mathematically simple and human interpretable models (rather than a black-box type approach) built over the descriptors, are generated and simplified. Consequently, descriptors (both experimental and theoretical) as well as mechanisms towards determining patterns of activity, selectivity, and stability towards OxR can be identified, thereby guiding electrocatalytic design. Our efforts also include an integration of these experimental and theoretical data via an easily accessible web-platform named cathub (https://www.catalysis-hub.org/). The platform includes modules with a command line interface to access and upload data. Currently, such integration of theory with experimental features like spectroscopic data, voltametric maps, diffraction patterns, catalyst characterization information etc. into an easily accessible online database for the field of heterogeneous catalysis is lacking. We therefore believe our approach and integrated database will be invaluable for the discovery of novel OxR catalysts.
On a bigger picture, ML extracted human-interpretable models along with derived physical insights can accelerate materials discovery and design as well as foster new collaborations through the sharing of ideas via easy access to online database of theory and experimental physicochemical properties.
References
1. Roling, L.T., L. Li, and F. Abild-Pedersen, The Journal of Physical Chemistry C, 2017. 121(41): p. 23002-23010.
2. Deo, S; Stenlid, J. H.; Abild-Pedersen, F. (In preparation).
3. Deo, S; Stenlid, J. H.; Abild-Pedersen, F. (In preparation).