(191m) Developing of Theoretical Methods for Multi-Metallic Alloys
AIChE Annual Meeting
2022
2022 Annual Meeting
Engineering Sciences and Fundamentals
Faculty Candidates in CoMSEF/Area 1a, Session 2
Monday, November 14, 2022 - 5:54pm to 6:06pm
The ultimate challenge in heterogeneous catalysis is to develop low-cost catalytic materials with high stability, activity, and selectivity. The discovery of novel catalytic materials is needed to solve various societal problems, such as global warming, producing value-added commodity, and ensuring a green and sustainable energy future. In this respect, bi-metallic and multi-metallic catalysts with complementary functionalities and unprecedented tunability have emerged with great opportunity to create tailored catalyst with desired functionalities [1-2]. However, making predictions about these material is challenging due to their immense structural and compositional complexity. Therefore, designing new computational strategies to accelerate the screening of optimum catalysts across vast compositional space is highly desirable.
Background
The chemisorption of molecular and atomic species on solid-state material surfaces is a central concept that is used to explain observed phenomena and estimated properties of new, tailored material surfaces [3]. Despite having made significant contributions to the physical description of chemisorption, the existing physics-based methods - including the Newns-Anderson model, the d-band model, and models based on coordination-numbers - remain inadequate for interpreting complex systems such as multi-metallic alloys (MMAs) [3-8]. These models typically consider the influence of the substrate surface on the adsorbate while overlooking the adsorbate-induced effects on the surface. Therefore, developing theoretical models to accurately predict the chemisorption as function of structure and composition is crucial to guide in the design of materials with desired properties. Whereas the current trend to harness the power artificial intelligence and big-data approaches have been increasingly successful in describing alloyed catalysts, there is a growing need for the development of physics-based, transparent, and generalizable models to rationalize chemisorption behavior.
Discussion on methods
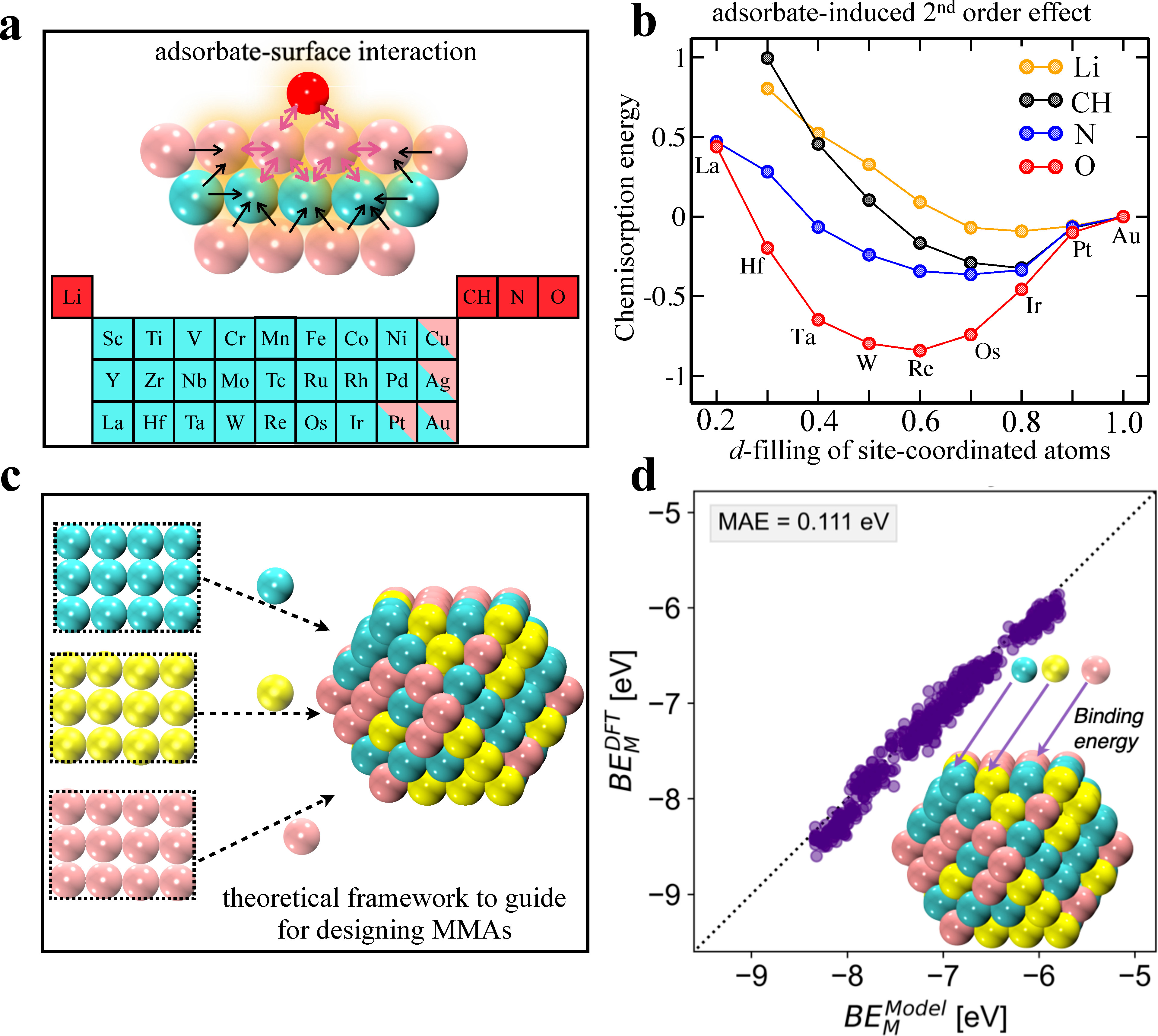
In this presentation, I will discuss two methods developed by me and collaborators. In the first method, we introduce a new theory-derived model for predicting chemisorption energies on surfaces of transition metal alloy. We integrate adsorbate-induced perturbations to the adsorption site with properties of the electronic d-band of the substrate. These perturbations interact with the chemical environment of the site where local variations yield different responses in the chemisorption behavior. These variations explicitly give rise to deviations from the typical linear behavior of the adsorption energy with electronic structure descriptors such as the d-band center. We find a mean absolute error of 0.13 eV on comparing the adsorption energies of various adsorbates (O, N, CH, and Li) on a wide variety of transition metal surface alloys to density functional theory-estimated values, highlighting the robustness of our approach. The model is fully transparent and is based on a site’s first and second d-band moments, as well as the d-band filling of the alloying atoms in first coordination shell surrounding the site [9]. This generalized theoretical model with additional physical insight and transparency provides the atomic-level guidance needed for designing complex alloys with desired catalytic properties.
In the second method, we design a simple and general strategy for predicting the site stability of MMA surfaces and nanoparticles spanning a wide-ranging combinatorial space. We parameterize the energy of constituent atoms as a function of their coordination number by performing a small handful of density functional theory (DFT) calculations of metal atom adsorption energies on clean and dilute bi-metallic alloy surface slabs. The obtained parameters are solely dependent on the identity and local coordination environment of a metal atom in both clean and diluted systems. By interpolating the obtained parameters from clean to completely diluted system, we can determine the stability of any given atom-site in any conceivable chemical environment across a wide range of morphologies, sizes, atomic compositions, and arrangements. We have validated our method across massive data sets by varying the elemental concentration of bi- and tri-metallic sets of Ir, Rh, Ru, Pd, and Pt and getting the high accuracy in predicting site stability, with errors between 0.11 and 0.15 eV. Our approach is highly efficient and easily transferrable to higher order of multi-metallic systems regardless of the percentage of the constituent elements. The approach only requires (N2 •16) DFT calculations for MMAs composed of N elements [10]. Our theoretical paradigm is robust in providing the atomic-level vision and accelerating the reverse engineering for emerging high entropy alloy catalysts.
References
1. Montemore, M. M.; Nwaokorie, C. F.; Kayode, G. O., Catalysis Science & Technology 2020, 10, 4467–4476.
2. Pedersen, J. K.; Batchelor, T. A.; Bagger, A.; Rossmeisl, J. ACS Catalysis 2020, 10, 2169–2176.
3. Hammer, B.; Nørskov, J. Chemisorption and Reactivity on Supported Clusters and Thin Films; Springer, 1997; pp 285–351.
4. Nørskov, J. K.; Studt, F.; Abild-Pedersen, F.; Bligaard, T. Fundamental concepts in heterogeneous catalysis; John Wiley & Sons, 2014.
5. Newns, D. Physical Review 1969, 178, 1123.
6. Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q. H.; Pohl, M. D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.;
Bandarenka, A. S. Science 2015, 350, 185–189.
7. Roling, L. T.; Choksi, T. S.; Abild-Pedersen, F. Nanoscale 2019, 11, 4438–4452.
8. Schmidt, J.; Marques, M. R.; Botti, S.; Marques, M. A. npj Computational Materials 2019, 5, 1–36.
9. Saini, S; Stenlid, J. H.; Abild-Pedersen, F. (under review).
10. Saini, S; Stenlid, J. H.; Abild-Pedersen, F. (under preparation).
Figure: (a) shows a general representation of the adsorbate-surface interaction. (b) Adsorption energy of O, N, and CH as a function of d-filling on Au NSAs having 5d metals in the second layer. All energies are relative to pure Au. (c) Schematic illustration to design the MMAs. (d) The predicted binding energies of atoms sites on MMAs surfaces are plotted against those obtained with DFT.