(491h) Atomistic Simulations of Ion Mobilities in Lithium-Ion Batteries
AIChE Annual Meeting
2022
2022 Annual Meeting
Engineering Sciences and Fundamentals
Lithium & Beyond: Fundamental Advances in High Performance Batteries I
Wednesday, November 16, 2022 - 2:30pm to 2:45pm
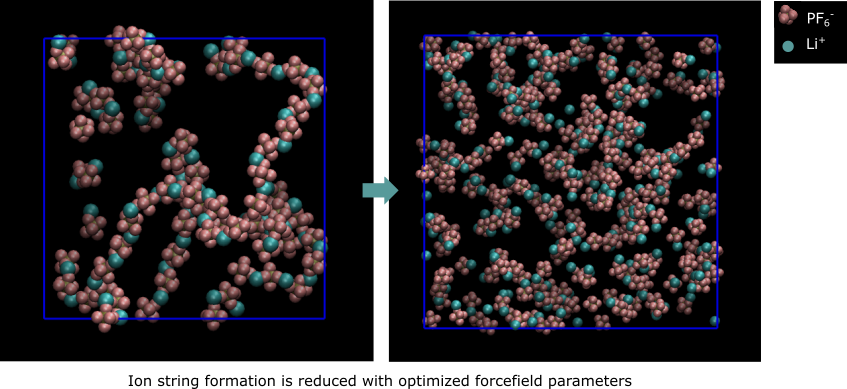
Simulations of transport properties for concentrated ionic solutions like lithium-ion battery electrolytes require accurate intermolecular forces. Standard OPLS potentials results in formation of large “ion stringsâ€, which drastically reduce ion mobility. But are these strings real, or an artifact of the potentials? Our group has recently developed efficient simulations for electrolytes of the osmotic equation of state, which we compare with experimental data to validate ion potentials. Excessive inter-ionic attraction results in string formation and reduced osmotic pressure. Using osmotic pressure is preferred over transport property measurements to validate the potentials as it is an equilibrium property, easily measured, and sensitively affected by formation of clusters. We have used validated forcefields to determine ion mobilities in solutions of Li+/PF6- in ethylene carbonate/dimethyl carbonate (EC/DMC). Our simulation approach applies forces to ions and measures drift velocities, thus measuring Onsager coefficients directly. The Onsager matrix can be converted to traditional Stefan-Maxwell diffusion coefficients, which have been inferred from experimental measurements on this system.