(527e) Determining Solvation Energies for Aqueous-Phase Reaction Adsorbates Using Ab-Initio Molecular Dynamic Simulations
AIChE Annual Meeting
2022
2022 Annual Meeting
Topical Conference: Material Interfaces as Energy Solutions
Interfacial Chemical Conversion
Wednesday, November 16, 2022 - 1:55pm to 2:10pm
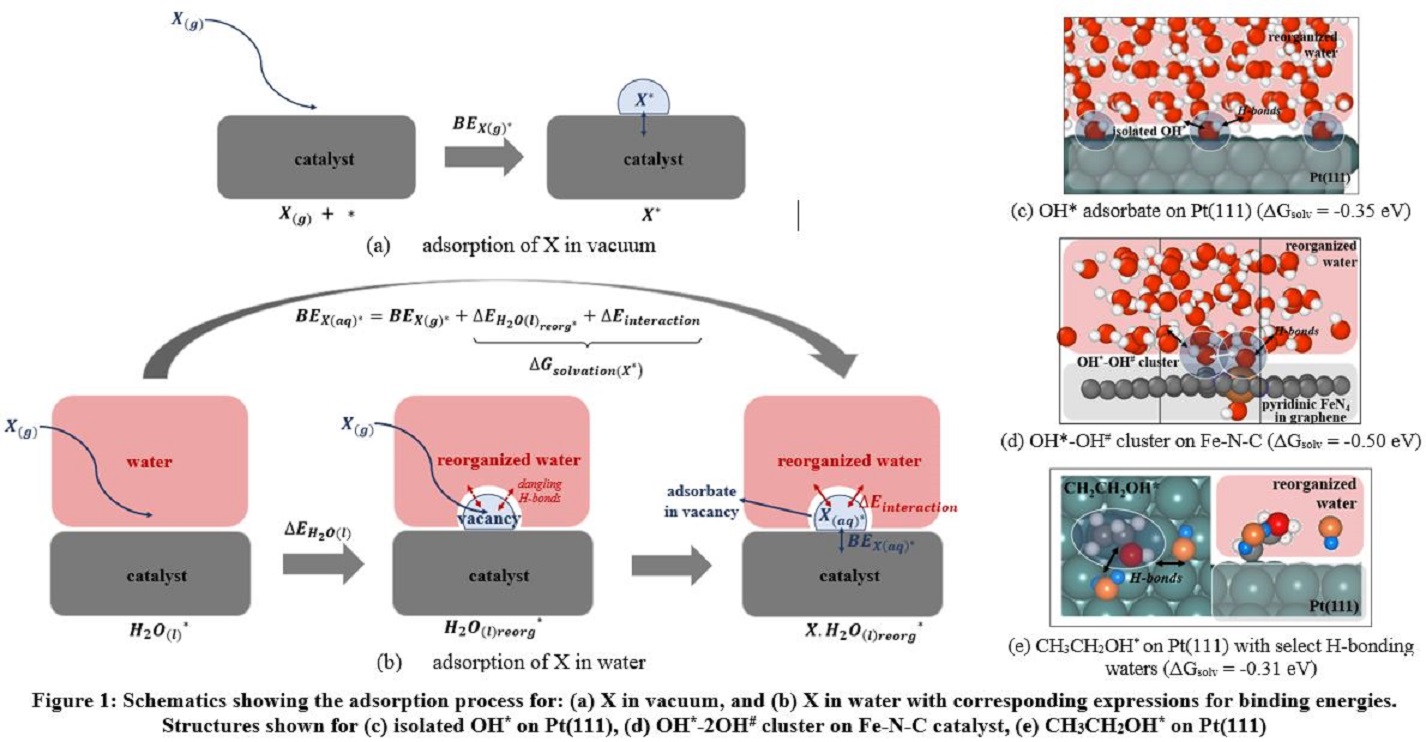
The application of this method is illustrated in the context of oxygen reduction catalysis on platinum and Fe-N-C catalyst surfaces. Among other observations, we find that OH* intermediates interact strongly with one another when clustering around hydrophilic FeN4 centers located in hydrophobic graphene surfaces. Interestingly, this behavior causes the water molecules to solvate such clusters of hydrophilic intermediates to a lesser extent than they solvate isolated intermediates, since these coadsorbates form intramolecular hydrogen bonds within the cluster, and the solvent effect is correspondingly less. In contrast, coadsorbed OH* and OOH* intermediates adopt quite different structures on the surface of hydrophilic Pt(111) in aqueous solvents, as compared to adsorption in vacuum, due to a comparable order of interaction energy with solvent water molecules compared to the coadsorbates. Irrespective of the affinity of the catalyst for water, hydrophilic adsorbates exhibit significant stabilizing solvation free energies that directly impact oxygen reduction catalysis on both Pt and Fe-N-C catalysts. We close by briefly demonstrating how the solvation energy scheme may be extended to more complex C1 and C2 intermediates with both hydrophobic alkyl groups and hydrophilic hydroxy groups, of relevance to the methanol and ethanol electrooxidation reactions, on Pt(111) and Pt(211) surfaces. The alkyl moieties in the intermediate have a destabilizing effect owing to non-favorable hydrophobic interactions with solvent water molecules, while the hydrophilic hydroxy groups, have a stabilizing effect with favorable H-bonds forming with the solvent molecules.