(55g) Co-Oriented Fluid Functional Equation for Electrostatic Interactions (COFFEE) for Mixtures: Phase Behavior and Fluid Structure in Mixtures of Differently Polar Fluids
AIChE Annual Meeting
2022
2022 Annual Meeting
Engineering Sciences and Fundamentals
Thermophysical Properties: Theory and Experiments for Charged Systems
Monday, November 14, 2022 - 10:00am to 10:20am
Using molecular simulation data of polar model fluids, the new perturbation theory Co-Oriented Fluid Functional Equation for Electrostatic interactions (COFFEE) [3] aims at the simultaneous description of dipolar and H-bonding interactions. COFFEE differs from classic perturbation theories in that it separates the polar contribution to the free energy into a near field and a far field contribution, the former describing intermolecular interactions between direct neighbors and the latter particles at greater distances. The far field contribution is formulated as in classic perturbation theories. Within the near field however, the polar interactions are described while directly considering the preferential orientations between molecules that result from the polarity. These preferential orientations are captured by the orientation distribution function (ODF), which may be used to describe mutual orientations for molecules with central and off-center dipoles alike. For this purpose, COFFEE’s near field parameters were fitted to reproduce ODF from molecular dynamics simulations. The direct incorporation of the orientational structure is a unique feature of COFFEE that also allows for the prediction of thermophysical properties that depend on intermolecular orientation such as the relative permittivity [4, 5].
COFFEE has thus far been limited to the description of pure fluids. In this contribution, the recent extension of COFFEE to mixtures is presented. This new extension includes both the near field and the far field contributions and thus enables the prediction of both the ODF and VLE in mixtures of model fluids ranging from completely nonpolar to strongly polar while also considering off-center dipoles. As the nonpolar component, the Lennard Jones (LJ) fluid is chosen, while varying the energy parameter to generate LJ fluids with different volatilities. The polar components are modeled by the Stockmayer (ST) fluid, which consists of a LJ center with a superimposed point dipole, and by the shifted Stockmayer (sST) fluid, for which the point dipole is shifted away from the dispersive center. Several different dipole moments are considered for both the ST and the sST fluid. Binary mixtures of these components are investigated for a wide range of temperatures and across the whole range of compositions. For this purpose, a large base of molecular simulation data is established, including both ODF and VLE data. The VLE were calculated using the molecular simulation tool ms2 [6] and subsequently, ODF were calculated in the single-phase regions using the molecular dynamics software ls1 mardyn [7].
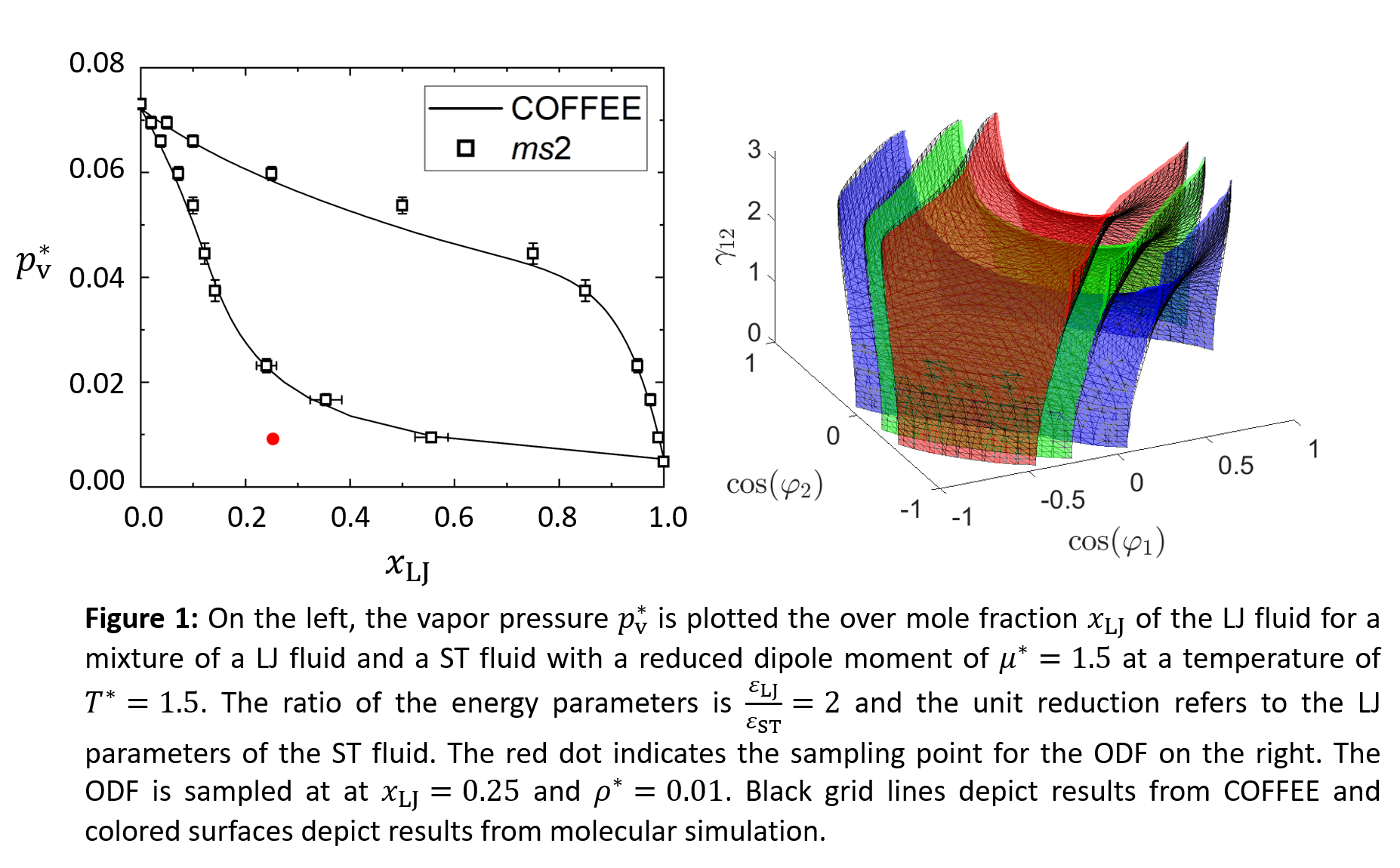
This large data base is then used to extend COFFEE to mixtures and assess the predictive ability of the theory. In a first step, the near field contribution is extended based on the original expression in a way that allows for the prediction of the ODF in mixtures without fitting mixture parameters. It is found that this approach leads to a satisfactory prediction of the ODF in mixtures with particularly accurate results in gas phases and for any binary mixtures containing one polar and one nonpolar component. For the prediction of VLE, good agreement between simulation and theory is found for a broad range of mixtures while keeping computational demand reasonably low. Figure 1 shows a VLE envelope and an ODF for a mixture of a LJ and a ST fluid as calculated by molecular simulation and with COFFEE. To characterize mutual orientations between two ST particles, three angles are required. φ1 and φ2 are the angles between the dipole vectors of the particles and the axis that connects the centers of mass of the particles. γ12 is the rotation angle around this axis. The ODF is illustrated in terms of isosurfaces that show mutual orientations that occur with the same probability. Orientations that occur with an average probability are shown as a green surface. The red surface represents mutual orientations that occur with a 40% higher frequency than those on the green surface and on the blue surface, orientations occur at a 40% lower frequency.
In this contribution, an excerpt of the molecular simulation results for ODF and VLE and the respective predictions from COFFEE are shown for a variety of mixtures containing ST and LJ fluids. The theory is critically assessed and compared to alternative approaches.
References
[1] E. Arunan, G. R. Desiraji, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci, D. J. Nesbitt: Defining the hydrogen bond: An account (IUPAC Technical Report), Pure Appl. Chem. 83 (2011) 1619-1636.
[2] K. Langenbach, C. Engin, S. Reiser, M. Horsch, H. Hasse: On the Simultaneous Description of H-Bonding and Dipolar Interactions with Point charges in Force Field Models, AIChE J 61 (2015) 2926-2932.
[3] K. Langenbach: Co-Oriented Fluid Functional Equation for Electrostatic interactions (COFFEE), Chem. Eng. Sci. 174 (2017) 40-55.
[4] K. Langenbach, M. Kohns: Relative Permittivity of Dipolar Model Fluids from Molecular Simulation and from the Co-Oriented Fluid Functional Equation for Electrostatic Interactions, J. Chem. Eng. Data 65 (2020) 980-986.
[5] M. Kohns, J. Marx, K. Langenbach: Relative Permittivity of Stockmayer-Type Model Fluids from MD Simulations and COFFEE, J. Chem. Eng. Data 65 (2020) 5891-5896.
[6] R. Fingerhut, G. Guevara-Carrion, I. Nitzke, D. Saric, J. Marx, K. Langenbach, S. Prokopev, D. Celný, M. Bernreuther, S. Stephan, M. Kohns, H. Hasse, J. Vrabec: ms2: A molecular simulation tool for thermodynamic properties, release 4.0, Comput. Phys. Commun. 262 (2021) 107860.
[7] C. Niethammer, S. Becker, M. Bernreuther, M. Buchholz, W. Eckhardt, A. Heinecke, S. Werth, H.-J. Bungartz, C. W. Glass, H. Hasse, J. Vrabec, M. Horsch: ls1 mardyn: The massively Parallel Molecular Dynamics Code for Large Systems, J. Chem. Theory Comput. 10 (2014) 4455-4464.