(682a) The Influence of Atomic-Scale Active Site Distributions on Selectivity in Electroreduction of CO2
AIChE Annual Meeting
2022
2022 Annual Meeting
Catalysis and Reaction Engineering Division
CO2 Upgrading III: From Fundamental to Applied CO2 Electrocatalysis II
Friday, November 18, 2022 - 8:00am to 8:18am
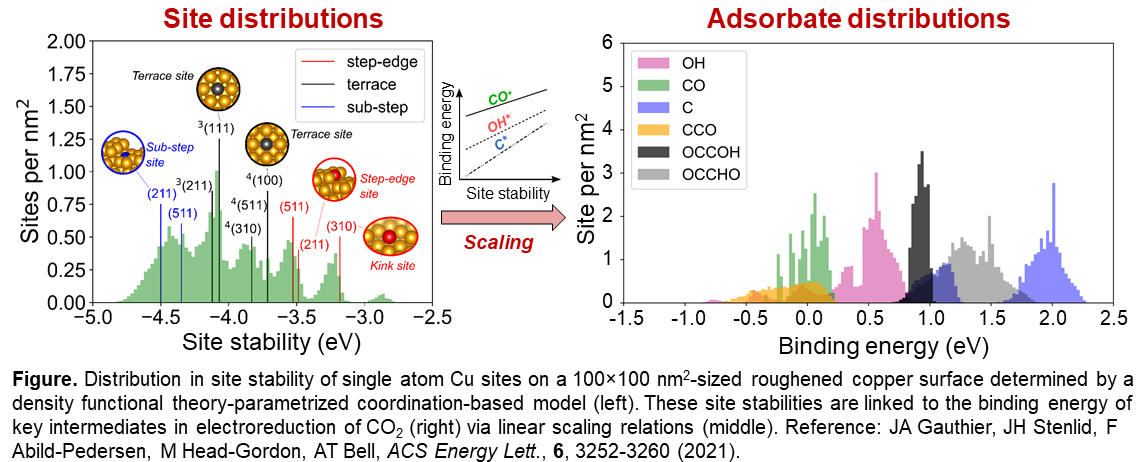
Roughened copper-based electrocatalysts surfaces are known to exhibit enhanced selectivity towards C2+ products during electroreduction of CO2. In this work, we investigate the atomic scale origin of this enhancement on nanofeatured copper surfaces. Model structures for rough surfaces of up to 100 x 100 nm2 size are obtained by using a carefully benchmarked effective medium potential and simulated sputtering. The relaxed structures are analyzed with a coordination-based stability model to obtain distributions of site stabilities of copper atoms in top, bridge, 3-fold, and 4-fold hollow sites. A broad distribution of sites is identified, and the stabilities deviate significantly from sites on idealized copper facets including common terrace, step, and kink surfaces. We thereafter establish linear relationships between the local site stability and the adsorption energy of key intermediates in the electrochemical CO2 reduction (CO2RR). This allows estimation of binding energy distributions for, e.g, CO*, OH*, C*, CCO*, and OCCOH* on the model surfaces. Again, these differs largely from the adsorption energies onto single crystal facets. By connecting the adsorption energies to thermodynamic models, we are able to evaluate the role of surface roughening on selectivity among C1 and C2 products during CO2RR. From this we conclude that roughened copper presents a plethora of catalytically active sites for CO2RR with largely varied selectivity towards different products. The study proposes that the challenge for designing improved copper catalysts lies in engineering ideal sites without simultaneously generating other sites that leads to unwanted by-products. It also highlights the need for careful experimentation when comparing to theory. On the basis of our findings, a new paradigm is envisaged in atomistic modeling of heterogeneous catalysts that accounts for active site distributions and the influence of proximate sites with complementary (or competing) functionalities. Through this, enhanced understanding can be obtained yielding improved guidance in catalysis design.