2022 Annual Meeting
(682c) Understanding the Dynamic Evolution of Atomically Dispersed Cu Catalyst for CO2 Conversion to Ethanol Using Integrated in Situ and Computational Approach.
Authors
Xu, J. - Presenter, Kansas State University
Patel, P., Argonne National Laboratory
Liu, D. J., Argonne National Laboratory
Liu, C., Argonne National Laboratory
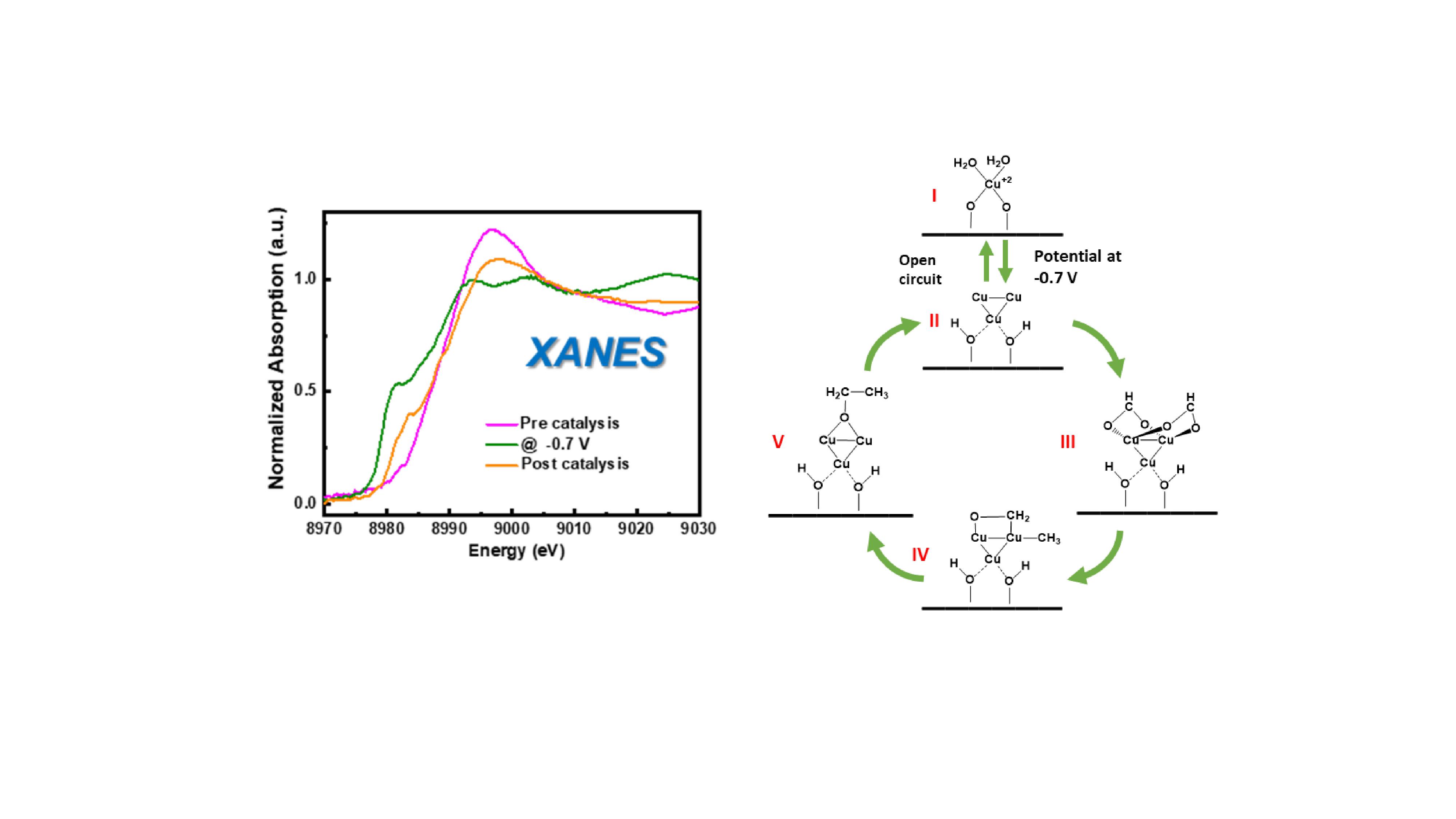
Direct electrochemical conversion of CO2 to ethanol offers a promising strategy to mitigate CO2 emissions while storing energy from renewable electricity. However, the high selectivity for producing C2+ chemical, such as ethanol, using current electrocatalyst is rather difficult. Our recent study reported that atomically dispersed copper (Cu) catalyst, prepared by amalgamated lithium metal approach, achieves a single-product Faradic efficiency (FE) of 91% at -0.7 V and the onset potential as low as -0.4 V for ethanol. Furthermore, by carrying out operando X-ray absorption near edge structure (XANES), we found a transformation from atomically dispersed Cu to Cu particles during the reaction. To gain further understanding of the dynamic evolution of the catalyst active sites, elucidate the active site structures, and investigate the CO2 reduction mechanism, a multiscale modeling approach combining density functional theory (DFT) calculations and theoretical XANES simulations were carried out. By matching theoretical and experimental XANES, we qualitatively revealed the structures of the pre, in-situ, and post catalysts, and we also confirmed the formation of large Cu particles and smaller clusters during electrocatalysis. Based on the mechanistic study by DFT, the limiting potential for CO2 reduction to ethanol is -0.41 V and and the rate limiting step is C-C bond formation with energy barrier as 0.58 eV. In addition, we also found that C-C bond formation energy barrier increases as particle size increases, thus the active centers that are responsible for CO2 reduction to ethanol are actually the smaller clusters, i.e. Cu3 and Cu6, with low C-C- bond formation energy barriers. Our computational results agree well with experimental investigation and validate our previous findings, which also provides guidance for future catalyst development.