2022 Annual Meeting
(684h) A Computational Workflow for Intermetallic Catalyst Discovery for Selective Hydrogenation Reactions
Authors
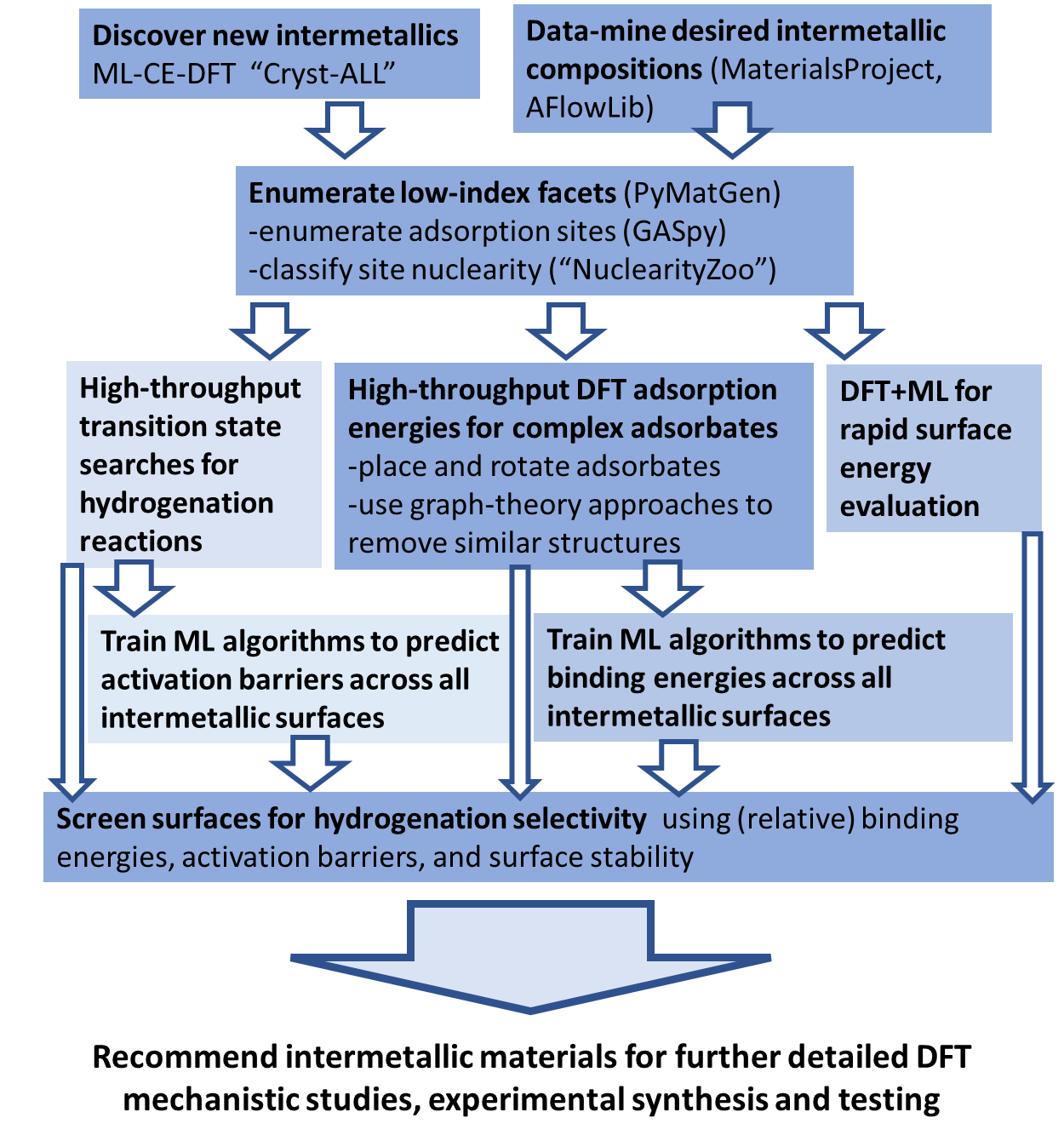
The development of large materials databases has made a large number of intermetallics structure and formation energies (computed with density functional theory (DFT)) available. We target combination of inert hosts (Zn, Cd, In, Ga, Al) with active late transition metals (Pd, Pt, Ru, Rh, Ag, Ir), and integrate a series of computational approaches that enumerate intermetallic bulk and surface structures, automate adsorbate placement, and screen for surfaces that show encouraging adsorbate/intermediate binding energies as promising selective hydrogenation catalysts.
The major result of this work is the series of tools that compromise the computational workflow used to identify intermetallics for selective hydrogenation reactions. Figure 1 illustrates this computational workflow incorporating the Cryst-ALL approach for rapid detection of the most stable bulk structures over a composition space and the enumeration of low index facets of these bulk structures with their active site nuclearities determined. An automated placement approach was developed in order to place complex key adsorbates on these low index facets to generate a database of DFT adsorption energies. This database becomes a training set for ML approaches, allowing for rapid expansion of adsorption energies to identify surfaces offering targeted binding properties.