(294f) Computer-Aided Molecular Design of Heat-Resistant Surfactants Using Classical Density Functional Theory
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Computational Molecular Science and Engineering Forum
Applications of Molecular Modeling to Study Interfacial Phenomena
Wednesday, November 8, 2023 - 9:15am to 9:30am
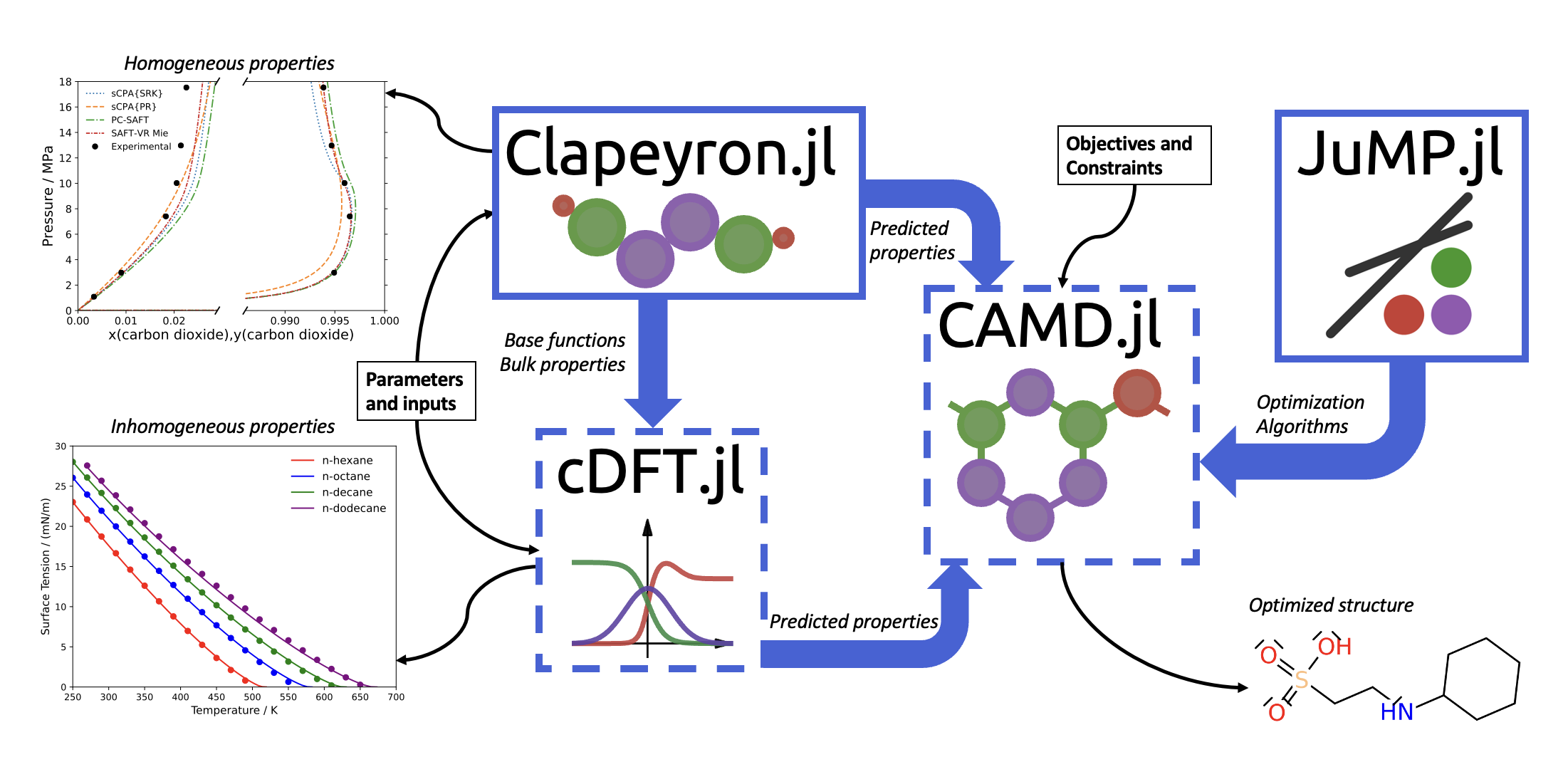
The investigation of innovative surfactants is a highly active research area, particularly regarding the identification of surfactants suitable for specific applications such as enhanced oil recovery. However, due to the scarcity of large-scale experimental databases, the implementation of machine-learning tools for the design of such molecules is a challenging task. Nonetheless, predictive models, such as classical density functional theory (cDFT), can be used in combination with a group-contribution methodology to overcome this problem, enabling the use of computer-aided molecular design (CAMD) frameworks. Therefore, in this study, we have developed open-source, generalized frameworks for both cDFT and CAMD. By implementing these tools, we utilized the well-established heterosegmented PC-SAFT functional coupled with mixed-integer non-linear optimization algorithms to design heat-resistant surfactants. The proposed molecules aligned with our anticipated structures for such surfactants, some of which have yet to be examined experimentally or through molecular dynamics simulations. Our intention is to expand our current framework to design anionic, cationic, and zwitterionic surfactants. Furthermore, these frameworks can also be extended to explore other design problems such as electrolytes for battery applications and solvents for carbon capture absorption.