(326g) Accelerating Materials Discovery for Electrochemical Reactions Using Iterative Machine Learning Strategies with Theoretical and Experimental Data
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Engineering Sciences and Fundamentals
Electrochemical Fundamentals: Faculty Candidate Session I
Monday, November 6, 2023 - 4:54pm to 5:06pm
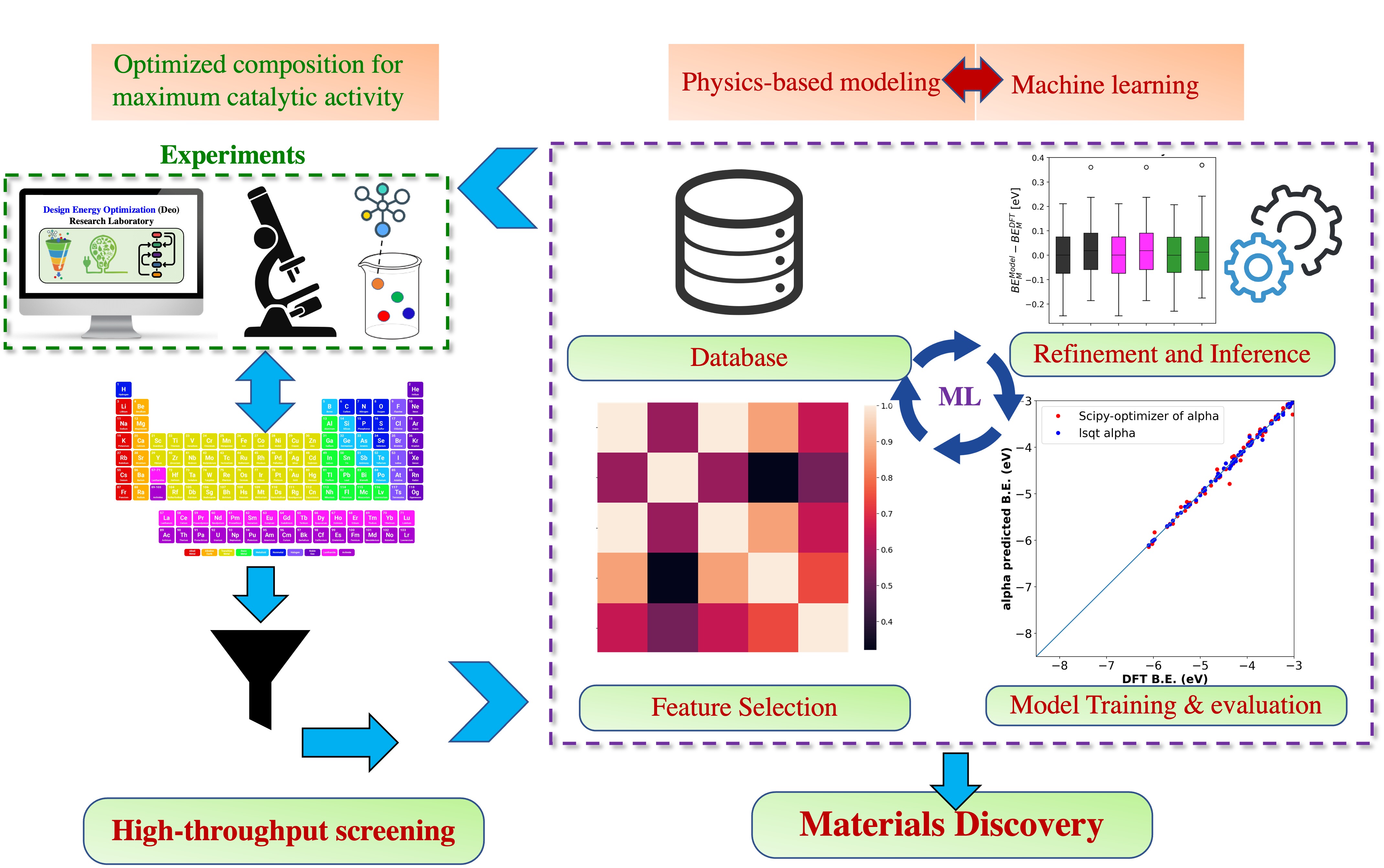
In my talk, I will demonstrate how the integration of physical models, high-throughput simulations, density functional theory (DFT), and ML can accelerate the process of materials discovery and catalyst design. Additionally, I will discuss how an iterative ML approach that utilizes both experimental and theoretical data can reveal insights on the relationship between structure and properties, enabling the prediction of experimentally relevant and stable ORR catalysts. This ML methodology offers benefits over ML strategies that rely solely on theoretically generated materials data. Furthermore, this strategy can also be applied effectively to other electrochemical reactions.
In particular, I discuss a high-throughput exploration and computation of materials of high interest for ORR, and their relevant properties (for ORR), based on a large number of bulk prototype materials including antimonates, oxides, pyrochlores, alloys, nitrides, and sulfides. Such effort is critical for broader, efficient use of fuel cell and water-splitting devices. The methodology leverages prototype DFT calculations to extract electronic and structural descriptors from bulk crystal structures of materials. This information is then integrated with experimental ORR catalyst data and machine learning (ML) techniques to efficiently identify the most relevant descriptors for making predictions under catalytically relevant conditions. Mathematically simple and human interpretable models (rather than a black-box type approach) built over the descriptors, are generated and simplified. Consequently, descriptors (both experimental and theoretical) as well as mechanisms towards determining patterns of activity, selectivity, and stability towards ORR has been identified, thereby guiding electrocatalytic design. The models produced have practical applications in transfer learning for predicting other active materials based on the derived mathematical correlations. Understanding the relationship between the onset potential for ORR and other descriptors allows for manipulation of these factors in catalytic materials, resulting in even larger sets of active materials. The study has identified experimental and theoretical descriptors, as well as mechanisms for determining patterns of activity towards ORR. Additionally, the models can be expanded to include similar materials such as sulfided (MSbSx) and nitrided antimonates (MSbNx) by incorporating universal electronic and structural descriptors. Similarities between extracted descriptors for M-O, M-N, and M-S would establish universality. Lastly, the models are benchmarked against standard ML methodologies (like gaussian regression models) and found to be accurate and transferable.
Our efforts also include an integration of these experimental and theoretical data via the CatHub (https://www.catalysis-hub.org/) Python API. The platform includes modules with a command line interface to access and upload data. Currently, such integration of theory with experimental features like spectroscopic data, voltametric maps, diffraction patterns, catalyst characterization information etc. into an easily accessible online database for the field of heterogeneous catalysis is lacking. We therefore believe our approach and integrated database will be invaluable for the discovery of novel OxR catalysts. On a bigger picture, ML extracted human-interpretable models along with derived physical insights from experimental data can accelerate materials discovery and design as well as foster new collaborations through the sharing of ideas via easy access to online database of theoretical and experimental physicochemical properties.