(576b) Biomimetic Strain-Stiffening in Fully Synthetic Dynamic-Covalent Hydrogel Networks
AIChE Annual Meeting
2023
2023 AIChE Annual Meeting
Materials Engineering and Sciences Division
Polymer Networks & Gels: Reprocessing & Vitrimers
Thursday, November 9, 2023 - 4:00pm to 4:15pm
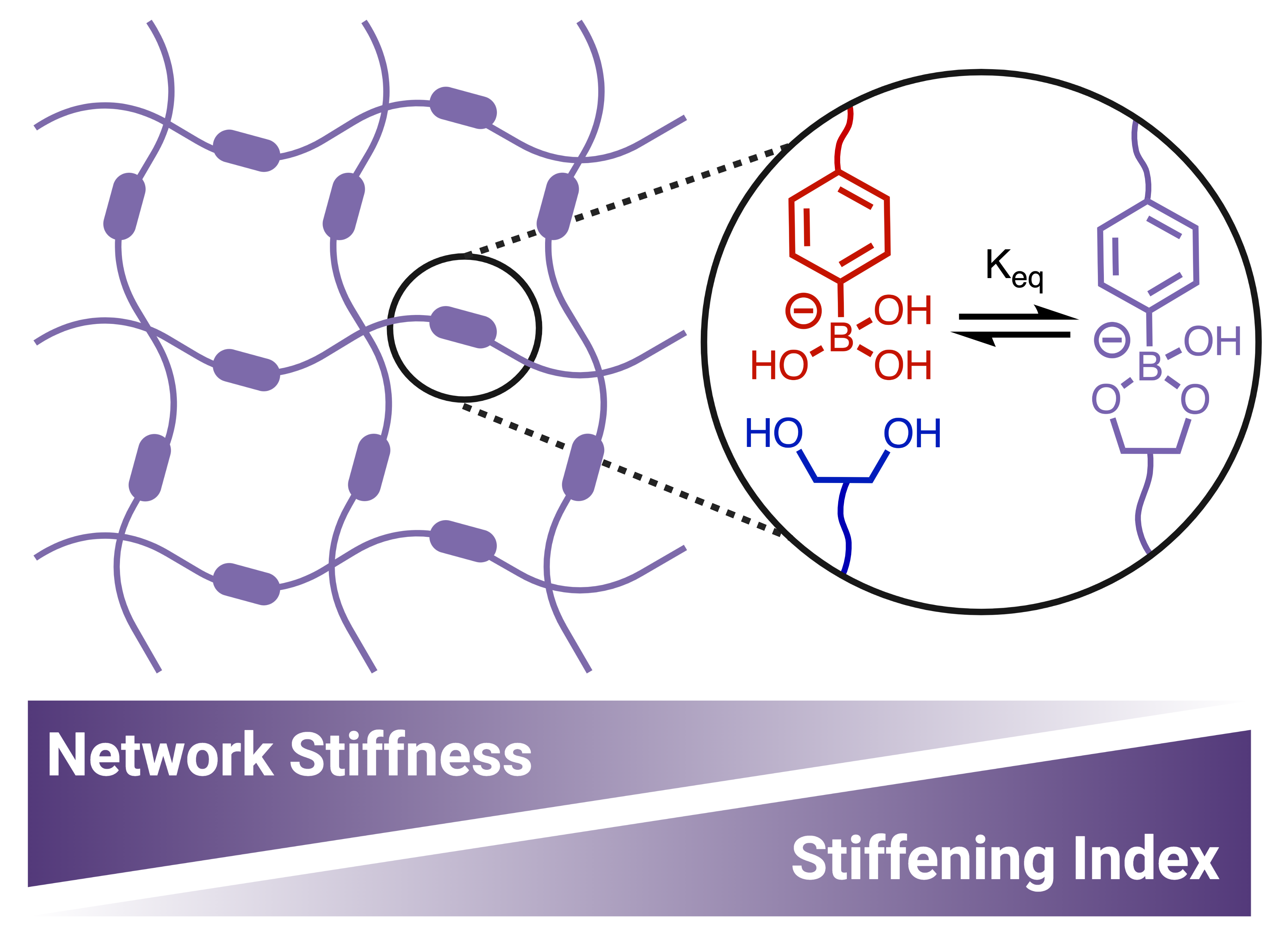
Mechanoresponsiveness is a ubiquitous feature of soft materials in nature; biological tissues exhibit both strain-stiffening and self-healing in order to prevent and repair deformation-induced damage. Strain-stiffening, a process by which materials become stiffer under applied deformation, is common among fibrous proteins such as actin, collagen, fibrin, and vimentin, and is often attributed to fibrillar structures which bundle and entangle. In recreating both the mechanical and structural features of soft biological tissues, hydrogels have been often explored for a number of biological and biomedical applications. However, synthetic polymeric hydrogels rarely replicate the mechanoresponsive character of natural biological materials, failing to match both strain-stiffening and self-healing functionality. Dynamic-covalent crosslinking imparts self-healing behavior, but it has been proposed that these dynamic, equilibrium-governed bonds are rarely strong enough to reach the stiffening regime caused by strain-induced elongation of polymer chains. As a result, this class of hydrogels rarely displays strain-stiffening. However, we have realized strain-stiffening behavior in fully synthetic ideal network hydrogels prepared from flexible 4-arm polyethylene glycol macromers via dynamic-covalent boronate ester crosslinks. Shear rheology reveals a strain-stiffening response in these networks as a function of polymer concentration, pH, and temperature. Increasing polymer concentration increases crosslink density while increasing polymer chain density proportionally. Due to the pH-dependence of boronate ester crosslinks, varying pH allows for the modulation of crosslink density while maintaining a constant polymer chain concentration. Temperature impacts the dynamics of the crosslinks, and thus the mechanical properties of the gels. Across all three of these variables, hydrogels of lower stiffness exhibit higher degrees of strain-stiffening, as quantified by the stiffening index. This is contrary to the relationship between gel stiffness and stiffening index that is typically displayed by strain-stiffening biopolymers. Whereas semiflexible biopolymer networks stiffen more acutely at higher concentrations and thus higher crosslink density, the ideal network PEG gels prepared here stiffen more dramatically at lower crosslink densities. The mechanism underlying this unusual stiffening response is attributed to a combination of entropic and enthalpic elasticity in these crosslink-dominant networks, contrasting with natural biopolymers that primarily strain-stiffen due to a strain-induced reduction in conformational entropy of entangled fibrillar structures. It is proposed that the increased stretch on individual chains results in an increased nonlinear elastic strain response in these gels. The reversibility and self-healing nature of the strain-stiffening response of these hydrogels is also evident upon strain-cycling, in which gels are subjected to periods of low strain and high strain repeatedly. Interestingly, when a high strain of 300% is applied, which is greater than the strain corresponding to the maximum value of G’ but less than the failure strain of the gel, a time-dependent decrease in modulus is observed, which is attributed to the reorganization of dynamic-covalent crosslinks under high strain. When the applied strain is decreased, a further, instantaneous decrease in modulus is observed, followed by a gradual recovery to the low-strain baseline. This behavior is thought to be due to the instantaneous relaxation of elastically active polymer chains, and the self-healing of the ruptured crosslinks on a slower timescale. Our work therefore offers key insights into crosslink-driven strain-stiffening in dynamic-covalent phenylboronic acid–diol hydrogels, and its dependence on experimental and environmental parameters. The biomimetic mechano- and chemoresponsive nature of this simple ideal-network hydrogel, as well as its biocompatibility and simple preparation, offers a promising platform for a variety of biologically relevant engineering applications.