(497h) Programming Structure-Function Relationships in Protein Megamolecules for Prodrug Cancer Therapy
AIChE Annual Meeting
2020
2020 Virtual AIChE Annual Meeting
Topical Conference: Chemical Engineers in Medicine
Precision Medicine and Cancer
Wednesday, November 18, 2020 - 9:45am to 10:00am
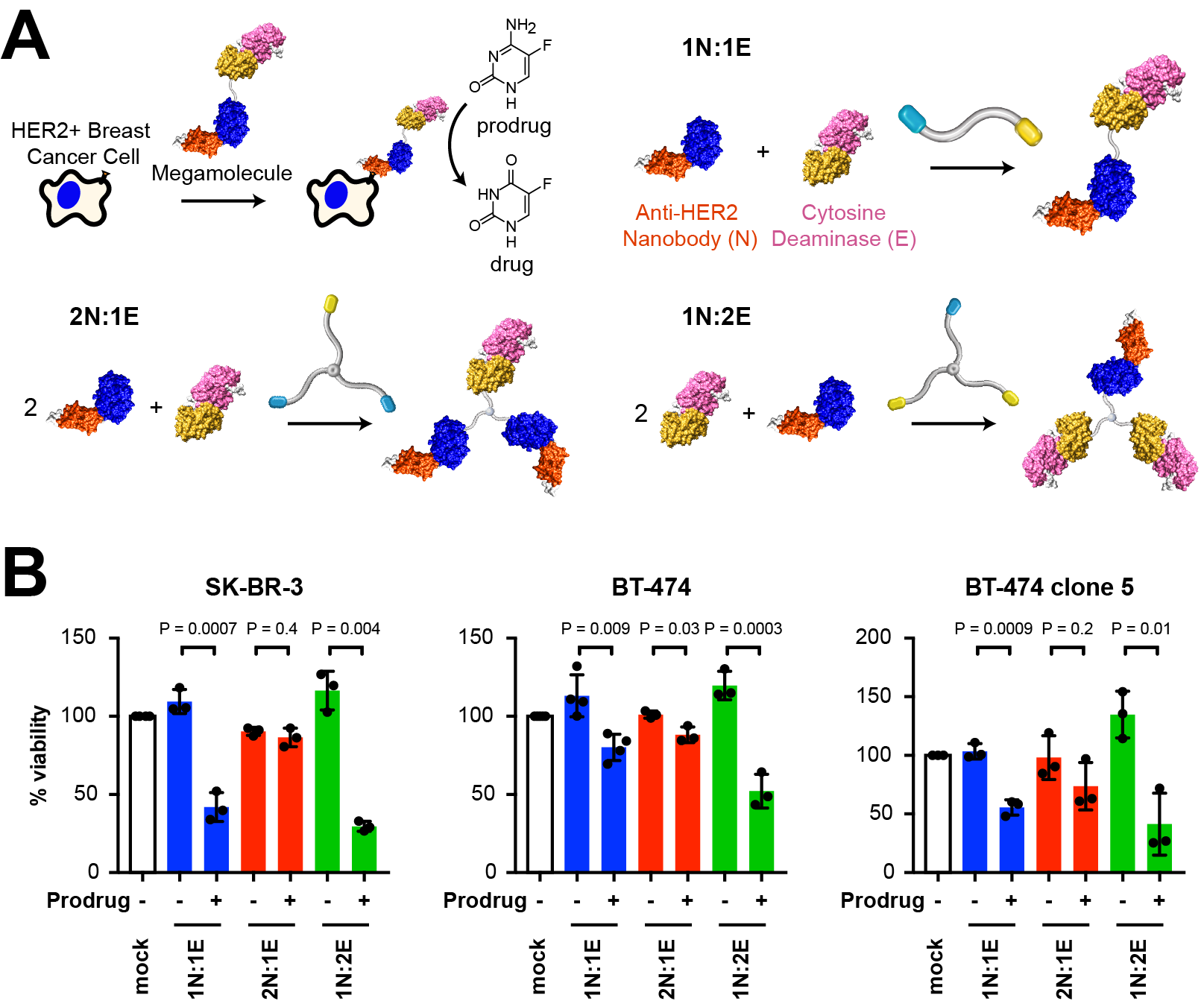
Antibody therapeutics have revolutionized the way we treat cancer due to their ability to specifically target tumors, leaving healthy cells unaffected. However, it is difficult to chemically modify the size, shape, valency, and structure of antibodies due to a lack of available tools. This limitation in programmability contributes to factors such as immunogenic responses, inability to penetrate solid-tumors, and high dosage requirements. Our lab has developed a protein assembly method known as the ‘megamolecule’ approach that can address this limitation. The megamolecule strategy provides precise, programmable control over therapeutic structures through enzyme mechanism-based covalent inhibition of fusion proteins containing a connector protein domain (cutinase or SnapTag) and an active protein, such as an antibody fragment. We used the megamolecule approach to create nanobody-enzyme conjugates for prodrug cancer therapy with precise control over both antibody and enzyme stoichiometry. As a first system, we used an anti-HER2 nanobody and an enzyme, yeast cytosine deaminase, that converts a nontoxic prodrug into a chemotherapeutic. We evaluated the colloidal behavior, antibody binding kinetics, enzyme activities for prodrug conversion, and cell-specific efficacy of the megamolecules in both adherent and spheroid culture. We found that increasing the number of enzyme domains within a molecule allowed for intramolecular dimerization of the yeast cytosine deaminase domains, resulting in a 7.5-fold improvement in the catalytic efficiency of the two-enzyme bearing protein therapeutic, as confirmed by enzyme assays and transmission electron microscopy. This improvement was followed by an increase in prodrug activation, resulting in a 4-fold cytotoxic improvement in adherent cell culture. Further, we generated fluorescent megamolecules with variable avidity to visually track and calculate the binding affinity of multi-domain nanobody megamolecules to HER2+ cancer cells in live cell culture using confocal microscopy. Our findings confirmed cell-specific binding and cytotoxicity of HER2+ cells compared to HER2- cells. We found that the addition of a second binding domain within the molecule resulted in a 5-fold reduction in the dissociation constant of the therapeutic for HER2. Studying the binding kinetics and solid tumor penetration of protein scaffolds using live-cell confocal microscopy has the potential to yield the design rules of cell targeting protein therapeutics. By enabling control over the specificity, orientation, and stoichiometry of active domains within a protein nanostructure, megamolecules represent next generation cancer therapies whose structure can be tailored for optimum activity.