
The fundamental thermodynamic equations developed for aqueous electrolyte systems provide a springboard for modeling the more-complex and less-understood mixed-solvent electrolytes.
A key enabling technology for developing and operating processes with electrolytes is first-principles-based process simulation, which requires accurate thermodynamic models. The first part of this two-part series, which was published in the March 2015 issue of CEP (1), introduced the fundamental thermodynamics of electrolyte systems and identified the critical thermodynamic parameters and the equations and relationships used to determine them. That article focused on aqueous electrolyte systems. This article builds on those concepts and discusses mixed-solvent electrolytes, which are more complex and not as well understood as aqueous electrolyte systems.
Mixed-solvent electrolyte systems
Electrolyte systems are prevalent in many industrial processes, playing leading roles in oil and gas production, natural gas sweetening, and CO2 capture and sequestration. Although aqueous electrolytes are the most well understood and discussed, mixed-solvent electrolytes are also important and deserve more attention. Examples of mixed-solvent electrolyte systems that contain a solvent in addition to, or instead of, water include:
- extractive distillation for anhydrous alcohol production, which uses salts to break water-alcohol azeotropes (2)
- organic amine solutions that remove H2S and CO2 from natural gas, and SO2 and CO2 from power plant fluegas (3)
- nitration processes that employ concentrated sulfuric acid to dehydrate and regenerate nitric acid solutions
- the HBr-Br2-H2O ternary system in H2-Br2 flow cells for energy storage
- ionic liquids (organic electrolytes that remain as liquid at room temperature) that are used as media for reactions and separations
- dissolved oils, polymers, and other organics that are present in fracturing fluids and saline flowback water from hydraulic fracturing operations (4).
Mixed-solvent electrolyte systems are much more complex than aqueous electrolyte systems and not as well understood. For example, because these systems contain multiple solvents, aqueous and nonaqueous, at high concentrations, several liquid phases may exist in equilibrium. Unlike in aqueous systems, liquid-liquid equilibrium must be considered in addition to vapor-liquid equilibrium. Very little is known about how these systems behave, e.g., how the electrolytes distribute between the liquid phases. Because of this lack of knowledge and the complexity of such systems, very few thermodynamic models have been proven to be applicable to mixed-solvent electrolyte systems.
Regardless of whether the electrolyte system is aqueous or mixed-solvent, the fundamentals of thermodynamic modeling remain the same. As discussed in the previous article on aqueous electrolytes (1), solution chemistry is the primary factor controlling the thermodynamic behavior of electrolyte systems. Solution chemistry accounts for partial and full dissociation, acid-base reactions, complex-ion formation, salt precipitation, and other reactions. Only after the solution chemistry has been properly accounted for can the other thermodynamic properties of interest be determined.
From a process simulation perspective, the most critical thermodynamic properties are the phase equilibrium properties, such as vapor pressure, gas solubility, liquid-liquid equilibrium, and salt solubility; calorimetric properties, such as liquid enthalpy and heat capacity; and speciation properties, such as pH and species concentrations. Among them, the speciation properties must be determined first, because speciation dictates the phase equilibrium behavior and calorimetric properties of electrolyte systems. For example, the solution chemistry determines the proportion of a strong acid, a weak electrolyte, or an ionic liquid that exists as molecular species or as ionic species. Such speciation concentrations, as well as activities, are essential for calculating reaction rates. The extents of speciation further determine the fugacities or the partial pressures of volatile weak electrolytes or strong acids involved in vapor-liquid equilibrium or liquid-liquid equilibrium. Moreover, the calorimetric properties of the solution depend primarily on the speciation, as the enthalpy of formation of ionic species is very different from that of the corresponding molecular species.
Note that thermodynamic properties of electrolyte systems are relative to a reference state. Two different reference-state conditions are used with electrolyte systems. The symmetric reference state, which defines the activity coefficient of a pure component as unity, is typically used to describe the activity coefficient of the solvent (e.g., water) at system temperature and pressure. In contrast, an unsymmetric reference state, which defines the activity coefficient of a component at infinite dilution as unity, is often chosen for electrolytes and molecular solutes, because the concentrations of these solutes are low relative to that of solvent water.
Basic thermodynamics
As stated previously, one of the first steps in modeling an electrolyte system is identifying the reactions taking place in solution. Consider an electrolyte system consisting of nitric acid, water, and magnesium nitrate. Nitric acid consumes one molecule of water and dissociates into a hydronium cation (H3O+) and a nitrate anion (NO3−):
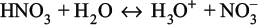
Separately, magnesium nitrate, which is a strong electrolyte, completely dissociates into a magnesium cation (Mg2+) and nitrate anions:
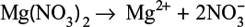
In a dilute aqueous solution of nitric acid, the nitric acid will almost completely dissociate. That solution can be treated as an aqueous electrolyte system, with water as the only solvent and undissociated molecular nitric acid as a molecular solute. As the concentration of nitric acid increases, the extent of nitric acid dissociation diminishes and the undissociated molecular nitric acid becomes a nonaqueous solvent — creating a mixed-solvent electrolyte system. Eventually, at very high concentrations of nitric acid, the extent of nitric acid dissociation is so low that most of the nitric acid exists in molecular form and the system becomes a nonaqueous electrolyte solution. The aqueous electrolyte thermodynamic framework is not applicable to systems with high concentrations of nitric acid.
Figure 1 shows the speciation reaction network in liquid-phase and vapor-liquid equilibrium of the nitric acid-water-Mg(NO3)2 ternary system. All of the chemical reactions and physical interactions between and among the species should be taken into account simultaneously when developing a thermodynamic model of the system.
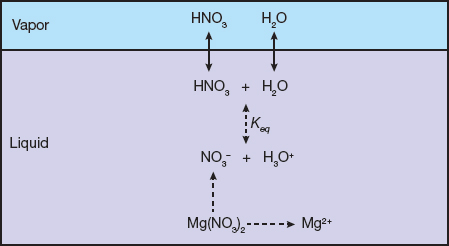
▲ Figure 1. It is important to identify and understand the reactions and speciation of a mixed-solvent electrolyte system. The nitric acid-water-Mg(NO3)2 ternary system involves several reactions and species in the liquid and vapor phases.
The first step toward representing the chemical reactions in this system is calculating the chemical equilibrium constants (Keq) for each reaction. These can then be used to determine the concentration of each species, and eventually the thermodynamic properties of the system.
To generalize the discussion, consider an electrolyte in the form of Mv+Xv– that partially dissociated into ν+ cations of M, each with a charge Z+, and ν– anions of X, each with a charge Z–(5):
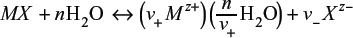
The chemical equilibrium constant for the partial dissociation reaction can be expressed in terms of the activity (ai) and activity coefficient (γi) of each component i:
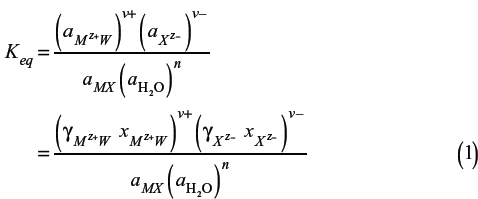
The concentrations and activity coefficients of each species can be expressed on a mole-fraction scale or a molality scale. While the molality scale is extensively used for aqueous electrolyte systems, molality-scale concentrations can approach infinity for concentrated electrolyte solutions. Therefore, it is more convenient to use mole-fraction-scale concentrations and activity coefficients for mixed-solvent electrolytes; the mole-fraction scale should be used for solvents, solutes, electrolytes, and ions in mixed-solvent electrolyte systems.
A reference state must be chosen for each species. The symmetric reference state is chosen for all solvents (i.e., pure liquid at system temperature and pressure). For ionic species, the unsymmetric reference state of aqueous-phase infinite dilution is preferred, mainly because extensive data are available for thermodynamic property constants of ionic species at this reference state. The choice of reference state should be considered and applied consistently for both the chemical equilibrium constant (Eq. 1) calculations and the activity coefficient calculations.
Phase equilibrium
The phase-equilibrium relationships used for aqueous electrolyte systems (Ref. 1, Eqs. 16–23) are applicable to mixed-solvent electrolyte systems. However, when more than one solvent is involved, two liquid phases may be present. The presence of multiple solvents and the complex molecule-molecule, molecule-ion, and ion-ion physical interactions in solution often result in rather nonideal phase-equilibrium behaviors that are best described by thermodynamic quantities such as chemical potentials, fugacities, and activity coefficients.
For vapor-liquid equilibrium, the chemical potentials (μi) of component i in the vapor and liquid phases are equal:

Alternatively, the equilibrium condition can be expressed in terms of fugacities (fi):

The fugacity of component i in the vapor phase is:

where φi is the vapor-phase fugacity coefficient of component i (which can be calculated with an equation of state such as the ideal gas or Redlich-Kwong equation of state), yi is the vapor-phase mole fraction, and P is the system pressure.
The fugacity of component i in the liquid phase is:

where γi is the liquid-phase activity coefficient, xi is the liquid-phase mole fraction, and fi0 is the liquid-phase reference fugacity.
The liquid-phase reference fugacity can be calculated from:

where pi0 is the saturation vapor pressure of component i at the system temperature, φi0 is the vapor-phase fugacity coefficient at the system temperature and pi0, and θi0 is the Poynting pressure correction from pi0 to the system pressure.
For liquid-liquid equilibrium, the equilibrium condition can be expressed in terms of the activities, i.e., the product of the liquid-phase activity coefficient (γi) and the liquid-phase mole fraction (xi), for liquid-phase I and liquid-phase II, since the liquid-phase reference fugacities cancel out.
For molecular species:

For electrolytes:

where γ± is the mean ionic activity coefficient and x± is the liquid-phase mole fraction of the electrolyte.
For volatile molecular solutes such as CO2, Henry’s law should be used to calculate the liquid-phase fugacity (1).
Thermodynamic models
The thermodynamic models for electrolyte systems are engineering expressions for excess Gibbs free energy [Gex] (or molar excess Gibbs free energy [gex]), from which activity coefficients and the other thermodynamic properties can be calculated. Three prominent thermodynamic models have been extensively used in process simulators for aqueous electrolyte systems: the Pitzer ion-interaction model, the OLI mixed-solvent electrolyte (MSE) model, and the electrolyte NRTL (eNRTL) model. The...
Would you like to access the complete CEP Article?
No problem. You just have to complete the following steps.
You have completed 0 of 2 steps.
-
Log in
You must be logged in to view this content. Log in now.
-
AIChE Membership
You must be an AIChE member to view this article. Join now.
Copyright Permissions
Would you like to reuse content from CEP Magazine? It’s easy to request permission to reuse content. Simply click here to connect instantly to licensing services, where you can choose from a list of options regarding how you would like to reuse the desired content and complete the transaction.